Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maria Grazia Giansanti and Version 2 by Catherine Yang.
The evolutionarily conserved target of rapamycin (TOR) serine/threonine kinase controls eukaryotic cell growth, metabolism and survival by integrating signals from the nutritional status and growth factors. TOR is the catalytic subunit of two distinct functional multiprotein complexes termed mTORC1 (mechanistic target of rapamycin complex 1) and mTORC2, which phosphorylate a different set of substrates and display different physiological functions. Dysregulation of TOR signaling has been involved in the development and progression of several disease states including cancer and diabetes.
- Drosophila
- mTOR signaling
- GOLPH3
- Golgi
1. Introduction
Target of rapamycin (TOR) is an evolutionarily conserved serine/threonine kinase, which functions as a central regulator of cellular growth, metabolism and survival in response to environmental cues including nutrients and growth factors [1][2][1,2]. Abnormal TOR signaling has been associated with several human diseases including cancer and diabetes [2][3][2,3]. The TOR kinase, belonging to the phosphoinositide 3-kinase (PI3K)-related kinase family, owes its name to rapamycin, a macrolide with potent antifungal activity discovered in a soil sample from the island of Rapa Nui (Easter Island) [4]. Subsequent studies of rapamycin revealed its immunosuppressive, antiproliferative and neuroprotective effects [5][6][7][5,6,7]. TOR was originally identified in the budding yeast Saccharomyces cerevisiae where mutations in two genes encoding TOR1 and TOR2 proteins confer resistance to the growth-inhibitory properties of rapamycin [8]. It was shown that rapamycin binds the intracellular cofactor propyl-isomerase, FK506 binding protein-12 (FKBP12), to inhibit the activities of TOR1 and TOR2 proteins [8]. Unlike yeast, most other species including humans and Drosophila melanogaster harbor only one gene encoding TOR, which is the catalytic subunit of two distinct functional multiprotein complexes termed mTORC1 (mechanistic target of rapamycin complex 1) and mTORC2 ([3][9][3,9]; Figure 1).
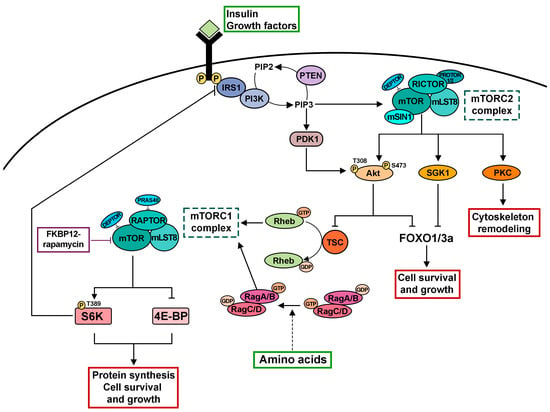
Figure 1. Cartoon depicting the mTOR signaling network. The mTOR kinase is the catalytic subunit of two distinct functional multiprotein complexes termed mTORC1 and mTORC2. mTORC1 and mTORC2 are characterized by a differential sensitivity to rapamycin and distinct accessory proteins. mTORC2 responds to growth factors by phosphorylating and activating several AGC family kinases including Akt, SGK1 and PKC, thereby regulating cell survival, cell growth and cytoskeletal dynamics. After binding of insulin or IGFs to their receptors and phosphorylation of the IRS1, PI3K is recruited to the cell membrane, leading to the accumulation of PIP3. PTEN functions as a negative regulator of this pathway converting PIP3 to PIP2. In turn, PIP3 triggers the recruitment of Akt to the plasma membrane followed by its activation via phosphorylation mediated by PDK1 and mTORC2. Akt phosphorylates the TSC2 subunit of the TSC complex to inhibit its GAP activity for the small GTPase Rheb, the direct activator of mTORC1. Amino acids regulate mTORC1 in a process that depends on the Rag family GTPases. A key function of mTORC1 is to promote protein synthesis for cell growth by phosphorylating S6K and 4E-BP. The following abbreviations are used: mTO RC1/mTORC2, mechanistic target of rapamycin complex 1/mechanistic target of rapamycin complex 2; FKBP12, propyl-isomerase, FK506 binding protein-12; LST8, lethal with SEC13 protein 8; RAPTOR, regulatory-associated protein of mTOR; PRAS40, proline-rich Akt substrate 40 kDa; DEPTOR, DEP domain containing mTOR interacting protein; RICTOR, rapamycin-insensitive companion of mTOR; mSIN1, mammalian stress-activated MAP kinase interacting protein 1; PROTOR1/2, protein-associated with rictor 1 or 2; AGC, cAMP-dependent, cGMP-dependent and protein kinase C; Akt, Protein kinase B; SGK1, serum- and glucocorticoid-induced protein kinase 1; PKC, Protein kinase C; IGFs, insulin-like growth factors; IRS1, insulin receptor substrate; PTEN, Phosphatase and TENsin homolog deleted on chromosome 10; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PIP2, phosphatidylinositol 4,5-Bisphosphate; PDK1, Phosphoinositide-dependent protein kinase-1; TSC, tuberous sclerosis complex; Rheb, Ras Homolog Enriched in Brain 1; S6K, ribosomal p70 S6 kinase I; 4E-BP, eukaryotic translation initiation factor 4E-binding protein.
Both complexes share the TOR kinase subunit and the mammalian homologue of LST8 (mLST8, lethal with SEC13 protein 8) [1][10][1,10]. However, they exhibit differential sensitivity to rapamycin and contain accessory proteins that are unique to each complex (Figure 1). The regulatory-associated protein of mTOR (RAPTOR; [10][11][10,11]), the proline-rich Akt substrate 40 kDa (PRAS40; [12][13][12,13]) and DEPTOR (DEP domain containing mTOR interacting protein; [14]) are associated with mTORC1. Conversely, mTORC2 is defined by the scaffolding protein rapamycin-insensitive companion of mTOR (RICTOR), which recruits the mammalian stress-activated MAP kinase interacting protein 1 (mSIN1; [15][16][17][15,16,17]), protein-associated with rictor 1 or 2 (PROTOR1/2; [18][19][18,19]) and DEPTOR [14] to form the complex. Many studies indicate that mTORC1 and mTORC2 play different physiological functions and have distinct substrates. mTORC1 controls cell growth and promotes protein translation by phosphorylating the downstream targets ribosomal p70 S6 kinase I (S6K) and the ribosome-associated eukaryotic translation initiation factor 4E-binding protein (4E-BP; [20][21][22][20,21,22]). On the other hand, mTORC2 regulates cytoskeletal remodeling, cell proliferation and survival by regulating the activity of members of the AGC (PKA/PKG/PKC) family of protein kinases [18][19][23][24][25][26][18,19,23,24,25,26]. Unlike mTORC1, mTORC2 was originally shown to maintain the ability to phosphorylate its substrates upon acute treatment with rapamycin and described as a rapamycin-insensitive mTOR complex ([18][19][18,19], Figure 1). However, it has been reported that prolonged treatment with rapamycin can impair mTORC2 integrity and activity by sequestering mTOR into rapamycin-bound complexes [27][28][27,28].
2. mTORC1 Signaling in Drosophila melanogaster Regulates Organ Growth
Extensive studies in genetically tractable organisms including Drosophila melanogaster have greatly contributed to understanding the intricate mTORC1 molecular pathway [9]. As a central cell growth regulator, mTORC1 responds to a variety of upstream signals including growth factors and the availability of amino acids and glucose [9]. mTORC1 is a downstream effector of growth factors such as insulin or insulin-like growth factors through the insulin receptor (InR)/phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway [29][38]. Following the binding of insulin or insulin-like growth factors (IGFs) to their receptors and phosphorylation of the insulin receptor substrate (IRS), PI3K is engaged by IRS in the cell membrane and converts phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). The PTEN tumor suppressor is a negative regulator of this pathway, acting as a phosphatase to convert PIP3 to PIP2 (for review, see [30][39]). In turn, PIP3 accumulation recruits and activates the serine-threonine kinase Akt (also known as protein kinase B) to the plasma membrane via phosphorylation mediated by the phosphoinositide-dependent protein kinase 1 (PDK1) and mTORC2 [9].
The PI3K/Akt pathway converges to mTORC1 via the key signaling regulator known as the tuberous sclerosis complex (TSC, [31][32][33][40,41,42]; Figure 1). The TSC complex, composed of TSC1 and TSC2 proteins, negatively regulates the mTORC1 pathway by functioning as a GTPase-activating protein (GAP) and a negative regulator of the GTPase protein Rheb (Ras homolog enriched in brain). The GTP-bound form of Rheb directly interacts with the kinase domain of mTOR and strongly stimulates its kinase activity [34][35][36][37][38][43,44,45,46,47]. AKT-mediated phosphorylation of TSC2 (also known as tuberin) disrupts the TSC complex, relieving its inhibitory effects on mTORC1 signaling [29][32][33][39][38,41,42,48]. The Tre2-Bub2-Cdc16 (TBC) 1 domain family, member 7 (TBC1D7) was recently identified as the third and stoichiometric component of the TSC complex, which stabilizes the association of TSC1 and TSC2 proteins by directly interacting with the TSC1 protein [31][40]. The near-atomic resolution of the human TSC complex by cryoelectron microscopy (cryo-EM) indicates that it has an arch-shaped structure with a 2:2:1 stoichiometry of TSC2 to TSC1 to TBC1D7 [40][49].
Genetic and biochemical characterization of the Drosophila homologs of the mTORC1 signaling components has illuminated the role of the mTOR pathway in organ growth and its relationship with the insulin pathway. Studies from two different research groups analyzed the phenotypes associated with mutations in the unique Drosophila TOR (dTOR) encoding gene, revealing striking similarities with the effects of nutrient deprivation or mutations in insulin signaling [41][42][50,51]. Mutational inactivation of the insulin/PI3K/Akt signaling pathway in Drosophila is associated with a decrease in cell size and organ growth accompanied by an extended G1 phase of the cell cycle, developmental delay and defective cell proliferation [32][41][42][43][44][45][46][47][48][49][50][51][52][53][41,50,51,52,53,54,55,56,57,58,59,60,61,62]. Like PI3K pathway mutants, animals carrying dTOR mutant alleles display defects in cell cycle progression and cell growth in several tissues during Drosophila development [42][51]. Loss of dTOR in Drosophila larvae also causes several cellular phenotypic defects that can be observed in amino-acid-starved wild-type larvae, including reduced nucleolar size, lipid vesicle aggregation in the larval fat body and a cell-type-specific growth arrest [42][51]. In imaginal disc cells from dTOR mutant larvae, the length of the G1 phase of the cell cycle is extended while the level of the G1/S-phase regulator cyclin E (CycE) is reduced. Moreover, the cell cycle arrest of dTOR mutant cells can be rescued by overexpression of CycE indicating that TOR signaling regulates cell proliferation by maintaining normal rates of CycE [42][51]. As in mammalian cells, dTOR is required for growth factor-dependent phosphorylation of activated Drosophila S6K (dS6K) and overexpression of dS6K in vivo can partially rescue the phenotypes of dTOR loss of function mutants [42][51]. The Drosophila genome harbors a single dS6K gene whereas the two orthologs RPS6KB1 and RPS6KB2 genes in humans encode the S6K1 and S6K2 proteins, respectively [54][55][56][57][58][63,64,65,66,67]. Most flies carrying null alleles of Drosophila S6K (dS6K) die before reaching the adult stage; the surviving flies are smaller than wild-type flies [59][68]. Notably, eye and wing tissues from mutant flies lacking dS6K exhibit a reduced cell size but a cell number that is comparable to wild-type tissues [59][68].
Characterization of mutations in the Drosophila Tsc1 (dTsc1) and dTsc2/gigas genes revealed that the heterozygosity can suppress the lethality associated with loss-of-function insulin receptor mutants [50][59]. Imaginal disc cells carrying mutations in either dTsc1 or dTsc2 show enhanced growth and increased size but retain normal ploidy [60][69]. Later studies demonstrated that Drosophila Tsc1 and Tsc2 proteins form a complex and antagonize insulin signaling to regulate cellular and organ size and function upstream of TOR [61][70]. Loss of dTsc1 and dTsc2 leads to a TOR-dependent increase in dS6K activity and inhibition of Drosophila Akt, with the latter effect that can be suppressed by loss of dS6K [61][62][70,71]. By using both in vitro and in vivo assays, Zhang and coauthors [38][47] provided evidence that the Drosophila small GTP-ase Rheb (dRheb) is the direct target of the Tsc2 GAP activity. Moreover, point mutations in the GAP domain of the Tsc2 protein disrupt the GAP activity towards dRheb. Consistent with these results, other studies in the same year showed that dRheb regulates cell growth, downstream of dTsc1-dTsc2 and upstream of dTOR in the insulin-dTOR signaling pathway [36][37][63][45,46,72]. In humans, somatic loss of heterozygosity in either the TSC1 or TSC2 tumor suppressor gene causes tuberous sclerosis, a disease characterized by the frequent development of benign tumors in various organs [64][65][73,74]. Although TBC1D7 has been reported as a core component of the TSC complex, loss-of-function mutations in TBC1D7 have not been reported in TSC patients [31][40]. However, germline homozygous truncating mutations in TBC1D7 have been associated with a rare autosomal-recessive megalencephaly and neurocognition syndrome [66][67][75,76]. These divergent phenotypes prompted Ren and coauthors [68][77] to use Drosophila melanogaster as a model system to investigate the physiological roles of Drosophila TBC1D7 (dTBC1D7) in vivo. In Drosophila, the major insulin-like peptides, ILP2, ILP3, and ILP5, are expressed in a set of median neurosecretory cells of the fly brain, known as insulin-producing cells (IPCs) and released in the hemolymph [69][70][78,79]. By using CRISPR/Cas9-mediated genome editing, Ren and coauthors [68][77] generated a null dTBC1D7 mutant. In contrast with TSC1 and TSC2 loss of function mutants, which are lethal, mutants lacking dTBC1D7 are viable, suggesting that the dTBC1D7 protein is not a constitutive component of the TSC. Indeed, they showed that dTBC1D7 is expressed in the IPCs of the fly brain and controls systemic growth in a cell-nonautonomous and TSC-independent manner, by selectively regulating the expression and release of ILP2.
Translationally controlled tumor protein (Tctp) is a highly conserved protein which plays an essential role in regulating cell growth and has been involved in tumorigenesis and tumor reversion [71][72][73][80,81,82]. Genetic and biochemical analyses in Drosophila have identified Tctp as a molecular component of the TSC-Rheb pathway required for controlling both the cell size and cell number in imaginal discs [74][75][83,84]. Work from Hsu and coauthors [74][83] showed that the Drosophila TCTP (dTctp) protein directly binds dRheb and functions as a guanine nucleotide exchange factor for it in both in vivo and in vitro assays. The cell growth defects associated with down-regulation of dTctp can be rescued by human TCTP (hTCTP), suggesting that the function of TCTP in the Rheb-mTORC1 signaling is evolutionarily conserved [74][83]. Consistent with this hypothesis, another study provided evidence in vitro that hTCTP can accelerate the GDP release of the human Rheb protein in biochemical assays [76][85].
The functional role of Drosophila PRAS40 (dPRAS40) in coupling insulin and mTORC1 signaling was investigated in vivo in flies carrying a loss-of-function mutant allele [77][86]. Biochemical and genetic analyses demonstrate that the dPRAS40 function regulates TORC1 activity and links insulin signaling to TORC1 in Drosophila [77][86]. However, unexpectedly, although PRAS40 is expressed in all tissues, it regulates TORC1 activity in ovaries, but not in other tissues, thereby influencing fertility but not animal growth. In mammals, 14-3-3 proteins were shown to regulate TOR signaling by binding several components of the mTORC1 complex including PRAS40 [78][87]. The Drosophila genome harbors two genes encoding 14-3-3 proteins, named 14-3-3 ε and 14-3-3 ζ, respectively [79][88]. In Drosophila 14-3-3 ε and 14-3-3 ζ proteins physically interact with both dTctp and dRheb proteins [75][84]. Single knockdown of either Drosophila 14-3-3 ε and 14-3-3 ζ in the wing or eye imaginal discs does not impair the wing/eye growth but causes genetic interaction with dTctp and dRheb to regulate organ growth. Moreover, double knockdown of 14-3-3 ε and 14-3-3 ζ disrupts organ growth and mTORC1 signaling by affecting the interaction between Tctp and Rheb [75][84].
A recent integrative analysis combined affinity purification-mass spectrometry with RNA interference screening and phosphoproteomic data, to identify new molecular targets of the InR/PI3K/Akt pathway in Drosophila [80][89]. This study revealed that approximately 10% of the interacting proteins are dynamically phosphorylated upon insulin stimulation. The Chaperonin containing TCP-1 (CCT) complex subunit CCT8 is one of the new molecular targets of insulin-dependent phosphorylation, identified during this proteomic analysis. The CCT complex has been predicted to control protein folding of about 10% of newly synthesized cytoplasmic proteins [81][90]. Consistent with the involvement of the CCT complex in InR/PI3K/Akt signaling, Kim and Choi [82][91] demonstrated that the Drosophila CCT complex physically interacts with dTOR, dRheb and dS6K proteins. Moreover, loss of the CCT complex results in severe cell growth defects during organ development and affects the level of phosphorylated dS6K while the total level of dS6K remains unchanged. Finally, the TOR signaling pathway regulates the transcription of the CCT complex. Overall, these results suggest that TOR-mediated control of cell growth also depends on the protein folding machinery.