Modulation of the human Ether-à-go-go-Related Gene (hERG) channel, a crucial voltage-gated potassium channel in the repolarization of action potentials in ventricular myocytes of the heart, has significant implications on cardiac electrophysiology and can be either antiarrhythmic or proarrhythmic. For example, hERG channel blockade is a leading cause of long QT syndrome and potentially life-threatening arrhythmias, such as torsades de pointes. Conversely, hERG channel blockade is the mechanism of action of Class III antiarrhythmic agents in terminating ventricular tachycardia and fibrillation. In recent years, it has been recognized that less proarrhythmic hERG blockers with clinical potential or Class III antiarrhythmic agents exhibit, in addition to their hERG-blocking activity, a second action that facilitates the voltage-dependent activation of the hERG channel. This facilitation is believed to reduce the proarrhythmic potential by supporting the final repolarizing of action potentials.
1. Facilitation of hERG Activation by Its Blocker
In response to membrane depolarization, hERG channels undergo slow activation followed by much more rapid inactivation
[1[1][2],
4], resulting in inward rectification in the current–voltage (IV) relationship with a maximal outward current at voltages between −10 and 0 mV. This gating property of hERG channels also results in a decreasing current with further depolarization, e.g., during the plateau phase of the ventricular myocyte action potential. The maintenance of this plateau is crucial for ensuring sufficient time for the entry of extracellular Ca
2+ into the myocyte and Ca
2+ release from the sarcoplasmic reticulum to enable cardiac contraction
[1,4][1][2]. As the myocyte repolarizes, hERG channel conductance increases due to recovering from inactivation and deactivation. The
IKr current during phase 3 repolarization of the ventricular action potential accelerates the repolarization and terminates the action potential. In experimental studies, the sigmoidal conductance–voltage (GV) relationship was analyzed by measuring tail currents at a negative voltage, where inactivation was weak. Many drugs interact with hERG channels and influence cardiac electrophysiology. Some drugs, known as blockers, reduce both the IV and GV relationships. Additionally, some hERG blockers not only reduce the IV and GV relationships but also shift them to the left, as shown in
Figure 1 [36,37,38,43,45,46,47][3][4][5][6][7][8][9]. This leftward shift in the GV relationship is referred to as the “facilitation” of voltage-dependent activation by the drug. In cases where the leftward shift was significant, the IV relationships of the control and drug-treated conditions may intersect, resulting in an increase in drug-induced hERG currents from the control at membrane voltages near the activation threshold. However, hERG channel activators increase hERG channel currents through a mechanism different from that of hERG blockers/facilitators (referred to as “hERG facilitators” in this review). Specifically, known activators enhance hERG channel activity by inhibiting inactivation
[39[10][11][12][13][14],
41,48,49,50], whereas the mechanism of hERG facilitation, which does not affect the inactivation process, differs from that of its activation. Perry had effectively summarized the pharmacological differences between the drugs in their review article
[42][15].
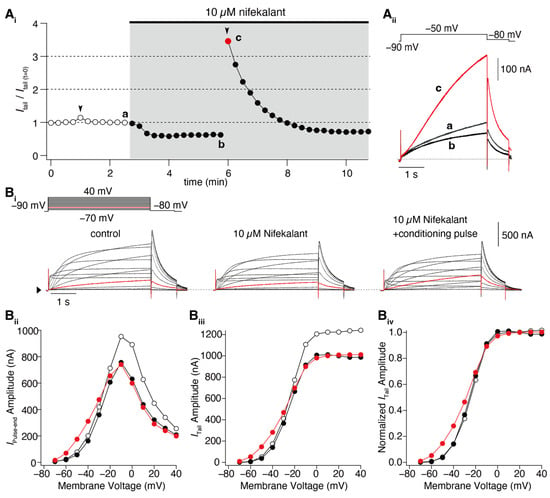
Figure 1. Nifekalant-induced facilitation of hERG activation. hERG channels were ectopically expressed in
Xenopus oocytes, and the currents were recorded using a two-electrode voltage clamp method. (
A) Depolarization-induced induction of facilitation. (
Ai) Time course of changes in hERG tail current (recorded at −80 mV) evoked by repetitive test pulses to −50 mV every 15 s. Conditioning pulses (+60 mV, 4 s) were applied twice in this experiment (black arrowheads), first in the absence and then in the presence of nifekalant. (
Aii) Superimposed cell currents recorded in the same oocyte before (Time (a) in (
Ai)) and after perfusion with 10 µM nifekalant, with (c) or without a conditioning pulse (b). Increased hERG current after the induction of facilitation effect by nifekalant is highlighted with red (red circle and trace in (
Ai) and (
Aii), respectively). (
B) Nifekalant plus conditioning pulse induced a shift in hERG activation curves. (
Bi) Representative traces of hERG currents in the control (left), block (center), and block/facilitation (right) conditions. (
Bii) IV relationship, (
Biii) GV relationship, and (
Biv) normalized GV relationship. Open, filled black, and filled red circles represent the control, 10 µM nifekalant without a conditioned pulse (block), and 10 µM nifekalant with conditioned pulse (block/facilitation) conditions, respectively. Panel (
A) was adapted with permission from Ref.
[37][4].
Numerous reports on hERG facilitators have been published
[36,37,38,43,45,46,47][3][4][5][6][7][8][9]. Among these hERG facilitators, the group focused on Class III antiarrhythmic agents such as amiodarone and nifekalant
[36,37,44,51][3][4][16][17]. These agents are highly effective in terminating refractory ventricular tachycardia and fibrillation and can induce voltage-dependent facilitation, as shown in
Figure 1. After treatment with nifekalant, the hERG currents induced by the test pulses decreased at −50 mV, which is considered a block and is thought to be the antiarrhythmic mechanism of Class III antiarrhythmic agents by prolonging the action potential duration and relative refractory period of ventricular myocytes. However, when strong depolarization to +60 mV was applied, the subsequent test pulse to −50 mV induced a large hERG current. This large hERG current was only observed in the presence of nifekalant, indicating that the drug (nifekalant) is necessary to increase the current. As shown in
Figure 1A
i, the effect of a single strong depolarization was transient. The response to the test pulses gradually decreased and eventually returned to the response observed before the application of strong depolarization. These transient changes in hERG currents were caused by a leftward shift in the GV relationship (
Figure 1B). A detailed biophysical assessment of this drug-induced leftward shift of the GV relationship revealed that both block and facilitation occurred with similar concentration dependence (
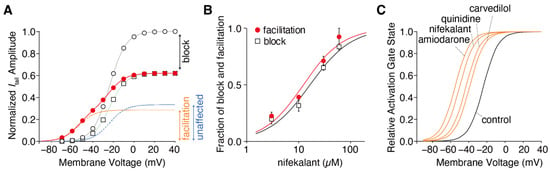
Figure 2). Additionally, the extent to which a drug shifts its GV relationship to the left is inherent in the drug itself. Notably, amiodarone shifted the curve by approximately 30 mV, whereas nifekalant shifted it by approximately 25 mV to the left (
Figure 2C). Although experiments are usually conducted using cells solely expressing hERG channels, a shift in the GV relationship also occurs when co-expressing the auxiliary subunit KCNE1
[44,52][16][18]. The expression system used did not significantly affect facilitation, even when recordings were carried out at different extracellular K
+ concentrations. Unlike activators, facilitators had little effect on inactivation.
Figure 2. Drug-dependent negative shift in the hERG activation curve. (
A) Voltage dependence of the hERG activation curves in the presence of 10 µM nifekalant. The tail currents of hERG in the absence (open circles) and presence of nifekalant with (filled circles) or without (open squares) the conditioning pulse were measured during the repolarizing pulse to −80 mV. The data were normalized to the current amplitude recorded following a voltage step of +10 mV in the absence of nifekalant. The model assumed two populations of channels with or without the facilitation effect of nifekalant (10 µM). The V
1/2 of activation for the facilitated fraction (orange dashed line) of channel was −50.7 mV, almost 28 mV negative to that of control channel (blue dashed lines). The red lines represent the double Boltzmann function (the sum of the Boltzmann functions for the facilitated fraction (orange dashed line) and the unaffected fraction (blue dashed lines)). (
B) Concentration–response relationships for compound-induced block and facilitation by nifekalant. (
C) Drug-dependent GV relationship in the facilitated fraction of hERG channels. Panel A was adopted with permission from Ref.
[37][4].
Certain voltage-dependent properties of the drug-induced facilitation aid the investigation of the mechanisms by which a drug can exert both hERG block and facilitation. Recently, the mechanism by which depolarization induces hERG facilitation was revealed. The voltage dependency of the induction of facilitation is associated with the voltage dependency of the hERG channel activation, specifically the opening of the activation gate in the pore
[51][17]. In this study, the D540K hERG mutant was utilized, which can be activated by both depolarization and hyperpolarization
[53][19]. In the wild-type hERG channel, facilitation is induced only by depolarization, whereas in the D540K hERG channel, it is induced by both depolarization and hyperpolarization stimuli
[51][17]. Furthermore, this study demonstrated that drugs can facilitate activation through hyperpolarization in the D540K hERG mutant
[51][17]. While the GV relationship of depolarization-induced activation shifted leftward, the GV relationship of hyperpolarization-induced activation shifted rightward. Considering the difference in structural changes in the voltage sensor domain caused by membrane depolarization and hyperpolarization, it is anticipated that drugs affect structural changes in the pore domain, facilitating the opening of the hERG channel pore when structural changes occur in the voltage sensor.
Nifekalant and other facilitators act as open-channel blockers for hERG. It is important to note that despite this, some readers may still question why depolarization (channel opening) only affects facilitation and not inhibition. The experimental protocol illustrated in Figure 1A utilizes 4 s long test pulses that are commonly used to stimulate slow-activating hERG channels. While the block also requires pore opening, this is not evident in the steady state following the channel opening. Furthermore, the blocking effect of the drugs was assessed by comparing the magnitude of inhibition with the current in the absence of drugs, which essentially evaluates the effect of the drug solely on the opened channels. This evaluation method makes it challenging to discern differences in inhibition at different membrane potentials. Conversely, the facilitation induced by the pre-pulse was evaluated, and the subsequent reopening by the test pulse did not typically cause significant pore opening. This double-pulse protocol enables clear observation of the voltage-dependent induction of facilitation.
2. A Possible Role of hERG Facilitation in hERG Block-Associated Arrhythmia
Yamakawa et al. investigated the facilitation effects of various non-cardiac drugs that block hERG channels
[43][6]. The results revealed that drugs such as fluoxetine (an antidepressant), haloperidol (an antipsychotic), and chlorpheniramine (an antihistamine) exhibit both conventional blocking effects on hERG channels as well as facilitation effects similar to antiarrhythmic agents, such as amiodarone and nifekalant. This suggests that the facilitation effect is not exclusive to antiarrhythmic agents and may be commonly observed among clinically used hERG blockers. This study also reported that some classical hERG blockers, including atenolol, terfenadine, and sotalol, did not exhibit facilitation
[43][6]. Terfenadine is an antihistamine that has been withdrawn from the market owing to its high risk of life-threatening arrhythmias. These findings suggest that the facilitation effect of hERG blockers may help reduce the risk of arrhythmias through their hERG-blocking mechanism. However, assessing the occurrence of facilitation in vivo presents technical challenges. In terms of the drug’s concentration, nifekalant can exert block and facilitate hERG channels in almost the same concentration-dependent manner
[37][4]. In addition, the repetitive excitation of ventricular myocytes may trigger facilitation. Experimental evidence has demonstrated that repeated stimulation with a voltage-clamp waveform that resembles action potentials can induce maximal facilitation
[44][16]. This may suggest that facilitation occurs in living hearts.
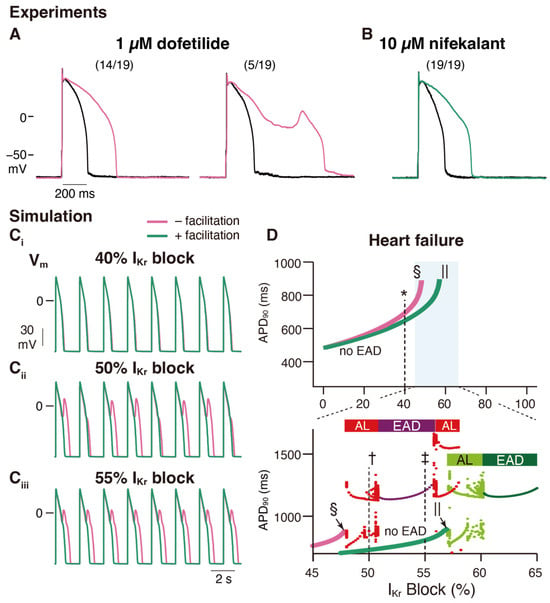
An in vitro validation was performed to examine the effects of nifekalant, a hERG blocker with facilitation, and dofetilide, a hERG blocker without facilitation, on rat ventricular myocyte action potentials. Even at concentrations that inhibited
IKr to a similar extent, dofetilide was more likely to induce early afterdepolarizations (EADs) than nifekalant, as shown in
Figure 53A,B
[44][16]. This suggests that facilitation may reduce the risk of arrhythmia. To further explore this concept, a theoretical study was conducted. The researchers developed a mathematical model to simulate facilitation and its impact on action potential waveforms
[44][16]. The facilitation model was formulated as a drug-induced shift in the GV relationship. By incorporating hERG block and facilitation models into a human ventricular myocyte action potential model (ORd human ventricular AP model
[73][20]), the researchers examined the influence of facilitation. Without a facilitation mechanism, increasing the concentration of a classical hERG blocker resulted in action potential prolongation and the development of EADs (
Figure 53C,D). However, in the presence of a facilitation mechanism, the prolongation of the action potential was suppressed, and a higher concentration was required to induce EADs (
Figure 53C,D). These observations theoretically illustrate that facilitation can lower the proarrhythmic risk associated with a drug.
Figure 53. Facilitation suppresses the development of early afterdepolarization related to
IKr blockade in ventricular cardiac myocytes. (
A,
B) Experimental study: Rabbit ventricular myocyte APs are more stable in nifekalant (
B) than in dofetilide (
A). AP responses in isolated rabbit ventricular myocytes were stimulated by minimal current injection (0.5 Hz) in whole-cell current clamp mode at 37 °C. Black line in (
A,
B) are control AP responses before the treatment. (
A) Representative AP responses without (left) or with (right) EAD in 1 µM dofetilide. Of the 19 cells treated with 1 µM dofetilide (magenta), five showed EAD responses (26%). Fourteen cells showed prolonged APD at 1 µM but did not show EAD responses. (
B) Representative AP responses to 10 µM nifekalant. All cells treated with 10 µM nifekalant (green) showed prolongation of APD upon treatment with 10 µM nifekalant but did not show EAD responses. (
C,
D) Simulation study: The effect of
IKr facilitation on APD prolongation and EAD development by
IKr block. (
C) Steady-state AP trains with 40% (
Ci), 50% (
Cii), and 55% (
Ciii)
IKr block in a heart failure model with and without facilitation. (
D) Effect of
IKr blockade and facilitation on APD and development of EADs in the heart failure model. Green and magenta lines indicate APD90 of AP (without EAD) for block with and without facilitation, respectively. The asterisk, dagger, and double-dagger indicate the conditions in (
C), respectively. The sections and pipes indicate the upper limits of the
IKr block, where APs are normally terminated. When EAD was observed, it was classified as either alternating or periodic EAD. In the bottom panel of D, red and deep purple dots indicate APD90 of AP with EAD for the block without facilitation, while light and deep green dots indicate APD90 of AP with EAD for the block with facilitation. Horizontal bars above the dots indicate alternating EAD, AL, periodic EAD, or EAD. This figure has been adapted with permission from Ref.
[44][16].
Class III antiarrhythmic agents are hERG blockers used clinically to suppress ventricular tachyarrhythmias
[1,31,32][1][21][22]. To suppress tachyarrhythmias without provoking
torsades de pointes, the
IKr block would ideally be use-dependent and prolong APD only in response to high-frequency stimulation
[7,74][23][24]. However, reverse frequency-dependent action on APs is a property common to Class III antiarrhythmic agents
[33,45,46[7][8][24][25][26][27][28],
74,75,76,77], and the associated proarrhythmic risk limits their clinical usefulness
[74,78][24][29]. The reverse frequency dependence of
IKr block was first explained by an increase in the slowly activated delayed-rectifier K
+ current with rapid heart rate
[75][26].
The mechanisms underlying these effects have also been analyzed
[44][16]. It is important to recall that the presence of blockers, such as nifekalant, can result in the crossing of the GV and IV relationships compared to their absence (
Figure 1 and
Figure 2)
[36,37][3][4]. When considering the trajectory of the ventricular action potential, depolarization occurs during phase 0 of the action potential, and this moves the myocyte membrane potential through the range of membrane potentials where facilitation increases the
IKr/hERG current. Consequently, the effects of facilitation were minimal during phases 0 and 2. As the action potential repolarization begins and the myocyte voltage returns to the range of membrane potentials where facilitation occurs, facilitation increases the
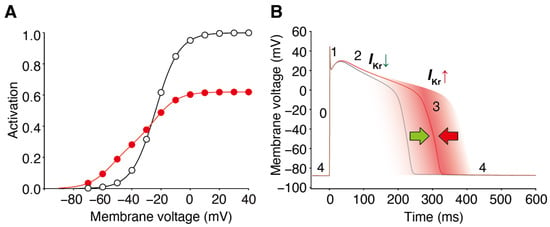
IKr/hERG currents. As a consequence, in this time- and voltage-window, the repolarizing currents are larger than in the absence of the drug. During a single ventricular action potential, hERG channel facilitators change their attributes to
IKr. It decreases the current first and then increases it later, which prevents excessive prolongation of the action potential and repolarization impairments (
Figure 64).
Figure 64. Block and facilitation impact on cardiac electrophysiology. (A) GV relationships of the control (open circles) and drug-treated conditions (red circles) intersect, resulting in drug-induced hERG/IKr currents decreasing at depolarized voltages and hERG/IKr currents increasing from the control at membrane voltages near the activation threshold. (B) The hERG channel blocker with a facilitation effect changes the attribute to IKr during the ventricular action potential. It decreases the current first and then increases it later, preventing excessive prolongation of the action potential and repolarization impairments. The different phases of the ventricular action potential (phases 0–4) are labeled. Green and red arrow indicate the dual drug actions on AP duration; IKr decrease by block prolongs (green), whereas IKr increase by facilitation prevents the prolongation of AP duration (red).
This facilitation-induced increase in the
IKr/hERG current during phase 3 can become even more pronounced when repolarization is delayed
[44][16]. This can be explained by the fact that, as the action potential duration is prolonged, the duration of the membrane potential within the range where facilitation occurs is also prolonged. This property can significantly affect the concentration-dependent and reverse frequency-dependent effects of hERG-blocking drugs on action potential duration.
Class III antiarrhythmic drugs are believed to exert their antiarrhythmic effects by extending the refractory period through hERG blockade, thereby prolonging the duration of action potential. There is some concern that the facilitation effect of hERG blockade on
IKr current might also affect the antiarrhythmic effects of Class III antiarrhythmics. However, simulations have indicated that this impact is minimal. The increase in
IKr current during phase 3 reduces excitability, offsetting the shortening effect on action potential duration. In simulations, prolongation of the relative refractory period by a hERG blocker with a facilitation effect was comparable to that of a classical hERG blocker
[44][16].