1. Toll-like LReceptor 4 Signaling as a Platform for Hepatocellular CarcinomaCC Vaccines
Cancer vaccines constitute a type of cancer treatment aiming to stimulate the immune system to target cancer cells differently as compared to what is applicable to vaccination against infectious diseases, where vaccines prevent disease development
[1][123]. There are several types of therapeutic cancer vaccines, including tumor cell vaccines, peptide vaccines, DNA vaccines, and dendritic cell vaccines
[1][123]. Recent scientific efforts have aimed to enhance their ability to target specific cancer antigens, induce long-term immune responses, and improve the efficacy of other cancer treatments such as chemotherapy, ICIs, and adoptive T cell therapy
[1][2][123,124]. In this direction,
toll-like receptor 4 (TLR4
) signaling could be exploited to improve HCC cancer vaccines
[3][4][125,126].
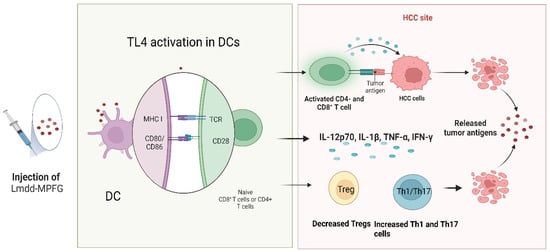
Wan et al. investigated the effectiveness of the Lmdd-MPFG vaccine in preventing tumor growth in C57BL/6 mice. The Lmdd-MPFG vaccine is based on an attenuated strain of the
Listeria bacterium, which has been modified to express a specific antigen called glypican-3 (GPC3), commonly found on the surface of HCC cells. The authors documented that Lmdd-MPFG activated TLR4 signaling in
dendritic cells (DCs
), leading to the upregulation of co-stimulatory molecules (CD80 and CD86) and the production of pro-inflammatory cytokines such as IL-12p70, IL-1β, TNFα, and IFN-γ. This enhanced the ability of DCs to prime antigen-specific CD4+ and CD8+ T cells and elicit an antitumor immune response. Furthermore, a decrease in the proportion of Treg cells and an increase in the percentages of Th1 and Th17 cells was documented. Overall, the authors suggested that TLR4 and NLRP3 signaling play a critical role in the immune regulation of DCs and the subsequent antitumor immune response
[3][125] (
Figure 1). Taking a step further, Silva et al. investigated the role of cold-inducible RNA binding protein (CIRP) as a cancer vaccine given the fact that ever-growing evidence links CIRP with liver carcinogenesis
[5][127]. CIRP was first identified as a protein that is upregulated in response to cold temperature stress, but subsequent research has shown that it is also induced by a variety of other stressors including heat shock, hypoxia, and inflammation. CIRP is a member of the RNA-binding protein family and is capable of binding to various RNA molecules, including messenger RNA (mRNA) and microRNA (miRNA). Through its interactions with RNA, CIRP can regulate gene expression by modulating RNA stability, translation, and splicing
[5][127]. Silva et al. documented that CIRP was overexpressed in HCC tumor tissues compared to adjacent non-tumor tissues. Additionally, they demonstrated that vaccination with ovalbumin linked to CIRP (OVA-CIRP)-based vaccine induced a potent immune response in vivo, resulting in increased production of cytotoxic CD8+ T lymphocytes (CTLs) and natural killer (NK) cells. Furthermore, the authors demonstrated that vaccination in combination with anti-PD-1 and anti-CTLA-4 ICIs led to enhanced anti-tumor activity and prolonged survival in a mouse model of HCC. This was associated with increased infiltration of CTLs and NK cells into the TME and decreased expression of immune checkpoint molecules
[4][126].
Figure 1. Basic principles of Lmdd-MPFG function. The vaccine uses an attenuated strain of Listeria modified to express GPC3 found on the surface of liver cancer cells. The vaccine activates DCs, leading to increased production of cytokines (IL-12p70, IL-1β, TNF-α, and IFN-γ), enhanced ability to prime T cells, and a decrease in Treg cells with an increase in Th1 and Th17 cells, promoting antitumor immunity. Created with
Biorender.com.
2. Toll-like Receptor LR4 Signaling as Regulator of Hepatocellular Carcinoma HCC Tumor Immune Landscape
TLR4 signaling plays an important role in the regulation of the immune landscape of HCC TME
[6][128]. TLR4 is expressed on various immune cells, including DCs, macrophages, and NK cells, as well as on HCC cells, activating several pathways such as NF-κB, MAPK, and phosphoinositide 3-kinase (PI3K)/Akt. TLR4 signaling can also promote the recruitment of immune cells in the TME, such as T cells and DCs, which can enhance the anti-tumor immune response. However, chronic activation of TLR4 signaling can also lead to the recruitment of immunosuppressive cells, such as Tregs and myeloid-derived suppressor cells (MDSCs), which can inhibit the immune response, promoting tumor growth
[7][129]. Therefore, the regulation of TLR4 signaling in HCC TME is complex and context-dependent.
2.1. Regulation of T and B lymphocytes by Toll-like Receptor 4 Signaling
2.1. Regulation of T and B lymphocytes by TLR4 Signaling
IL-22 and IL-22-binding protein (IL-22BP), are significantly upregulated in human HCC tissues as compared with non-tumor liver tissues, and the high expression of IL-22BP is associated with poor prognosis in HCC patients. Overall, the IL-22/IL-22BP axis plays a critical role in the development and progression of HCC, stimulating the proliferation and migration of cancer cells, and inhibiting apoptosis
[8][130]. TLR4 has been shown to play a critical role in the regulation of the immune response mediated by antigen-presenting cells (APCs). In more detail, TLR4 activation on APCs has been found to promote the expression of B7-H1, which, in turn, polarizes the differentiation of naïve T cells into Th22 cells, a subtype of CD4+ T cells that produce IL-22
[9][131]. Similarly, TLR4 plays an essential role in the recruitment and activation of Tregs in the liver parenchyma. The activation of TLR4 on macrophages increases the production of IL-10 and C-C Motif Chemokine Ligand 22 (CCL22) that promote the expansion and activation of CD4+CD25
highFOXP3+ Tregs, suppressing immune responses against the tumor. The inhibition of TLR4 signaling in macrophages reduced the number and activity of Tregs and slowed tumor growth in mice with HCC
[10][132]. Along the same line, the TLR4/C-X-C motif chemokine ligand (CXCL)10/C-X-C motif chemokine receptor (CXCR3) signaling axis, regulates Treg infiltration after liver transplantation. Specifically, the activation of TLR4 by LPS induces the secretion of CXCL10 by Kupffer cells. CXCL10 further binds to CXCR3 on Tregs promoting their migration to the liver and facilitating the establishment of an immunosuppressive microenvironment that promotes tumor growth and recurrence after transplantation
[11][133]. Finally, Wang et al. demonstrated that TLR4 signaling is required for the metabolic reprogramming of naïve CD4+ T-cells by NETs. Despite the lower absolute number of CD4+ T-cells in NASH, they documented that there was a selective increase in Tregs. The depletion of Tregs had a significant inhibitory effect on NASH-related HCC initiation and progression. Additionally, there was noted a positive correlation between increased hepatic Treg levels and NETs. RNA sequencing data revealed that NETs have an impact on gene expression profiles in naïve CD4+ T-cells, particularly those genes involved in mitochondrial oxidative phosphorylation. NETs promoted the differentiation into Tregs, facilitating mitochondrial respiration. TLR4 was needed for metabolic reprogramming of naïve CD4+ T-cells by NETs. In vivo blockade of NETs using
Pad4−/− mice or DNase I treatment reduced Treg activity. Targeting this process could provide a way to prevent liver cancer in patients with NASH
[12][134].
Xiao et al. further demonstrated that TLR4 signaling exerts significant influence in the regulation of B cells
[13][135], as they reported that PD-1
high B cells were significantly enriched in tumor tissues compared to blood, and expressed high levels of the immunosuppressive IL-10, TGF-β, and PD-L1, inhibiting the activation and proliferation of T-cells in vitro. They further demonstrated in vivo that the depletion of PD-1
high B cells in the HCC mouse model resulted in reduced tumor growth and improved OS. The generation of the PD-1
high B cell population is dependent on TLR4-mediated upregulation of B-cell lymphoma 6 (BCL6), which is reversed by the IL-4-induced phosphorylation of STAT6
[13][135].
2.2. Regulation of Dendritic Cells by Toll-like Receptor 4 Signaling
2.2. Regulation of DCs by TLR4 Signaling
DCs play a critical role in anti-cancer immunity. DCs are antigen-presenting cells that capture and process antigens and then present them to T cells to initiate an immune response. In cancer, however, the TME can inhibit the function of DCs, leading to a failure of the immune system to recognize and attack the tumor. Tumor cells may release factors that suppress DC function, such as TGF-β and IL-10, or hinder their differentiation and maturation, such as VEGF, ultimately leading to the prevention of T cell activation and ineffective immune responses
[14][136]. Yamamoto et al. demonstrated that the alpha-fetoprotein (AFP) inhibited the function of DCs, highlighting that AFP suppressed the production of pro-inflammatory cytokines IL-12p35 and IL-12p40 by DCs and impaired their ability to activate NK cells, which are important immune effector cells
[15][137]. These findings indicated that AFP could potentially impede the TLR-3 or TLR-4 signaling pathway, leading to the inhibition of mRNA translation of the
IL-12 gene
[15][137].
2.3. Regulation of Neutrophils by Toll-like Receptor 4 Signaling
2.3. Regulation of Neutrophils by TLR4 Signaling
In HCC, neutrophils can have both tumor-promoting and tumor-inhibiting effects. On the one hand, neutrophils can release various pro-inflammatory cytokines and chemokines that promote tumor growth, invasion, and angiogenesis, and suppress the activity of immune cells that are responsible for tumor surveillance, while, οn the other hand, they can demonstrate anti-tumor effects, as they can directly kill tumor cells by releasing toxic molecules such as ROS and NETs
[16][67]. Neutrophils can also promote the activation of cytotoxic cells to potentiate tumor immunity
[17][68].
Recent evidence implicates TLR4 signaling in the regulation of neutrophil function in HCC. TLR4 signaling plays a critical role in promoting the formation of NETs in HCC cells, which intriguingly contributes to the promotion of tumor growth and metastasis. The activation of TLR4/NF-κBp65/COX-2 signaling in HCC cells trapped by NETs promotes angiogenesis and tumor cell migration, inducing a pro-metastatic phenotype. As mentioned above, DNAse1 inhibitors, hydroxychloroquine (HCQ), and COX-2 inhibitors could suppress the induction of this pro-inflammatory response
[18][64]. In the same direction, Zhan et al. proposed a mechanism whereby HBV infection induces the expression of S100A9, a calcium-binding protein, which activates the TLR4/RAGE signaling pathway in CD66b+ neutrophils leading to the production of ROS and the release of NETs. Overall, the elevated levels of NETs facilitate tumor growth and metastasis, promoting angiogenesis and inducing an immunosuppressive microenvironment
[19][65].
2.4. Regulation of Myeloid-Derived Suppressor Cells by Toll-like Receptor 4 Signaling
2.4. Regulation of MDSCs by TLR4 Signaling
MDSCs are a heterogeneous population of immature myeloid cells that play a critical role in the immune evasion of tumors, including HCC
[20][138]. MDSCs exert their suppressive activity through various mechanisms, including the production of ROS, arginase, and cytokines that inhibit the function of T cells, NK cells, and DCs
[21][139]. MDSCs also promote the expansion of Tregs, which further suppress the immune response
[22][140] and limit the efficacy of checkpoint blockade
[23][141].
Accumulating evidence implicates TLR4 signaling in the regulation of MDSCs in HCC. In a study of 331 patients with HCC who underwent liver transplantation, those with a graft weight ratio (GWR) less than 60%, which is the weight of the liver graft divided by the standard liver weight of the recipient, had a higher risk of tumor recurrence compared to those with a GWR of 60% or more. Additionally, patients with a GWR of less than 60% or tumor recurrence had significantly elevated levels of MDSCs and CXCL10/TLR4. In a model of hepatic I/R injury plus major hepatectomy (IRH) in knockout mice lacking
Cxcl10 or
Tlr4 genes, monocytic MDSCs were significantly reduced while granulocytic MDSCs were not affected. Interestingly, deficiency of CXCL10 led to a decrease in the accumulation of TLR4+ monocytic MDSCs, and CXCL10 increased MDSC mobilization in the presence of TLR4. Moreover, matrix metallopeptidase (MMP) 14 was found to be the crucial molecule connecting CXCL10/TLR4 signaling and MDSC mobilization. Overall, evidence suggested that during the acute phase of injury, monocytic MDSCs were mobilized and attracted to the liver graft, which subsequently led to the promotion of HCC recurrence following transplantation. To reduce the likelihood of liver tumor recurrence after transplantation, a possible therapeutic approach could be the targeting of MDSC mobilization through CXCL10/TLR4/MMP14 signaling
[24][142]. Additionally, Hu et al. demonstrated that MDSCs accumulated in the liver during HCC development and contributed to tumor progression by suppressing the function of DCs. Specifically, the MyD88-NF-κB signaling in MDSCs caused IL-10 secretion, which led to the downregulation of IL-12 secretion by DCs, resulting in the suppression of T cell activation and proliferation due to the lack of co-stimulatory and major histocompatibility (MHC) class II molecules. In summary, TLR4 plays a crucial role in the upregulation and expansion of MDSCs, which contribute to HCC development by impairing the function of DCs and suppressing the activation of tumor-specific T cells
[25][143].
2.5. Regulation of Macrophages by Toll-like Receptor 4 Signaling
2.5. Regulation of Macrophages by TLR4 Signaling
Macrophages play a critical role in the development and progression of HCC. Macrophages can be classified into two main subtypes: M1 and M2 macrophages
[26][144]. M1 macrophages are involved in the initiation of the immune response and are characterized by the production of pro-inflammatory cytokines such as IL-1β, IL-6, and TNFα, while M2 macrophages are involved in the resolution of inflammation and tissue repair and are characterized by the production of anti-inflammatory cytokines such as IL-10 and TGF-β
[27][145]. In HCC, macrophages can promote or inhibit tumor growth depending on their phenotype and activation status. M2-like macrophages, which are characterized by the expression of markers CD163 and CD206, have been shown to promote tumor growth and metastasis by producing growth factors and cytokines that stimulate angiogenesis and immunosuppression; M1-like macrophages, on the other hand, which are characterized by the expression of markers CD86 and HLA-DR, have been shown to exhibit anti-tumor activity by producing cytokines that activate T cells and induce tumor cell apoptosis. The above are better analyzed elsewhere
[26][144] and a more in-depth discussion goes beyond the scope of
thisour re
searchview.
TLR4 signaling in macrophages is of paramount importance for the development of steatohepatitis-induced HCC
[28][146]. Miura et al. demonstrated in a mouse model of steatohepatitis-related HCC that mice lacking TLR4 had significantly fewer liver tumors than the control mice. They further showed that TLR4-expressing macrophages contributed to the development of HCC in
Pten-deficient (
PtenΔhep) mice by promoting the production of pro-inflammatory cytokines such as IL-6 and TNFα, as well as ROS. This is an important finding, as it suggests a potential therapeutic target for both the prevention and treatment of HCC in patients with NASH
[28][146]. The serine/threonine kinase 4 (STK4) protein is a critical regulator of TLR pathways in macrophages. Specifically, STK4 inhibits TLR4 signaling in macrophages, leading to a reduction in the production of pro-inflammatory cytokines
[29][147]. M1 polarization could be a promising therapeutic strategy against HCC, and mounting evidence in this direction has started to emerge. Astragaloside IV inhibits HCC progression by modulating macrophage polarization, and Min et al. showed that Astragaloside IV through the downregulation of the TLR4/NF-κB/STAT3 signaling pathway stimulates the aggregation of M1 macrophages and hinders M2 macrophages in vitro and in vivo
[30][148]. Furthermore, Pan et al. reported that combining TLR4 agonists with glucocorticoid-induced TNF receptor (GITR) agonists can reverse the M2 polarization of macrophages in HCC and enhance anti-tumor immunity. In murine HCC models, treatment with the LPS and DTA-1 (an agonistic GITR antibody) combination therapy significantly inhibited tumor growth compared to treatment with either LPS or DTA-1 alone. The combination therapy also increased the infiltration of M1 macrophages and CD8+ T cells in the TME, indicating enhanced anti-tumor immunity
[31][149]. Regarding the role of TAMs in HCC, Yao et al. showed that TAMs promoted the migration and EMT of HCC cells, which was mediated by the TLR4/STAT3 signaling pathway. They also found that inhibition of this pathway could reduce the effects of TAMs on HCC cells and thus may represent a potential therapeutic target for HCC
[32][80]. Evidence from another study also suggested that HCC-derived extracellular vehicles (EVs) containing prostate androgen-regulated transcript 1 (PART1), a long non-coding RNA (lncRNA), promoted the M2 polarization of macrophages and enhanced HCC cell proliferation and migration either through the PI3K/AKT or the Janus kinase (JAK)/STAT signaling pathway, suggesting that targeting these EVs may be a potential therapeutic strategy for HCC by inhibiting macrophage polarization and disrupting the tumor-promoting microenvironment
[33][94]. Further research is necessary to validate these findings and determine the potential clinical implications of targeting these signaling pathways.
2.6. Regulation of Fibroblasts by Toll-like Receptor 4 Signaling
2.6. Regulation of Fibroblasts by TLR4 Signaling
HCC mostly develops within a fibrotic microenvironment, where HSCs and carcinoma-associated fibroblasts (CAFs) play a crucial role in HCC progression
[34][150]. CAFs are a subset of fibroblasts that are present in the TME of HCC, and they partake in promoting tumor growth, progression, and metastasis through a variety of mechanisms. One of the primary functions of CAFs in HCC TME is the production and deposition of ECM components, creating a supportive environment for tumor growth and invasion
[35][151]. Moreover, CAFs can also regulate angiogenesis by producing various pro-angiogenic factors, including VEGF, FGF, and PDGF, which can stimulate endothelial cell proliferation and migration, leading to the formation of new blood vessels. Recent studies have also shown that CAFs in HCC TME can promote immunosuppression and escape from immune surveillance, and that they can produce factors that inhibit the activity of immune cells, including T cells, NK cells, and DCs, leading to a more permissive environment for tumor growth
[36][152]. Increasingly compelling evidence suggests that TLR4 signaling is implicated in the regulation of fibroblast/CAF-HCC cell interconnection.
Song et al. aimed to investigate the role of TLR4 signaling in the development of liver fibrosis and cancer in mice with hepatocyte-specific
Tak1 deletion
[37][105]. In more detail, this deletion leads to liver inflammation and fibrosis, which can eventually progress to liver cancer. They also reported that TLR4 and TLR9 activation is increased in the liver tissue of mice with
Tak1 deletion, and that this activation is necessary for the development of fibrosis and cancer
[37][105]. Similarly, Lie et al. proposed that LPS-induced differentiation of hepatic progenitor cells into myofibroblasts contributes to the development of HCC
[38][153]. In more detail, they documented that LPS exposure led to the differentiation of hepatic progenitor cells into myofibroblasts in vitro. In vivo, however, they reported that mice treated with LPS developed a more severe liver injury, increased myofibroblast activation, and a higher incidence of HCC
[38][153]. In addition, Lu et al. suggested that LPS promotes angiogenesis in HCC by stimulating HSC activation via the TLR4 pathway
[39][117], and they showed that LPS treatment increased HSC activation, as demonstrated by the increased expression of α-SMA and collagen I in HSCs. LPS treatment also led to increased angiogenesis in HCC, as evidenced by the increased microvessel density and VEGF expression
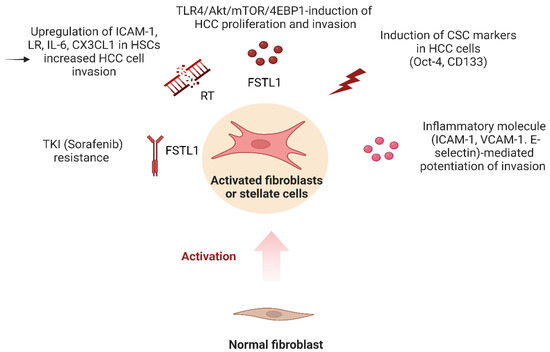
[39][117]. Loh et al. demonstrated that TLR4 signaling is critical in maintaining the immature phenotype of tumor-initiating cells (TIC)
[40][85]. Follistatin-like 1 (FSTL1) has been identified as a pro-inflammatory mediator in various fibrosis-related and inflammatory diseases. FSTL1 lineage cells give rise to myofibroblasts, and high FSTL1 expression in fibroblast activation protein (FAP)+ fibroblasts, was significantly associated with advanced disease in HCC patients. Treatment of Hep3B, HepG2 cells, and patient-derived 3D organoids with recombinant FSTL1 or conditioned medium collected from HSCs or cells overexpressing FSTL1 promoted HCC growth and metastasis by binding to the TLR4 receptor and activating AKT/mammalian target of rapamycin (mTOR)/4E-binding protein 1 (4EBP1) signaling. Blocking FSTL1 in a preclinical mouse model mitigated HCC malignancy and metastasis, sensitized HCC tumors to sorafenib, prolonged survival, and eradicated the TIC subset. These findings suggested that TLR4 and FSTL1 may serve as novel diagnostic/prognostic biomarkers and therapeutic targets for HCC
[40][85]. Ding et al. further stated that HCC cells acquire a pro-inflammatory and stem cell phenotype which is thought to play a crucial role in tumor initiation and progression
[41][154]. Specifically, they found that the presence of MRC-5 cells induced the expression of cancer stem cell markers in HCC cancer cells, including CD133 and Oct-4, and also upregulated the expression of several adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin
[41][154]. Finally, Shen et al. provided evidence that radiation exposure can promote the invasive potential of HCC cells through TLR4-mediated activation of HSCs
[42][155]. They reported that co-culture of MHCC97-L, Hep-3B, and LM3 cells with irradiated HSCs, significantly improved the invasion potential of HCC cells. Moreover, co-culture with irradiated HCC cells increased the invasive capacity of HSCs associated with activation phenotype and upregulated TLR4 signaling, linked to enhanced ICAM-1, laminin receptor (LR), IL-6, and CX3CL1 expression, and decreased the expression of toll-interacting proteins. Epigallocatechin-3-gallate (EGCG) was also found to inhibit TLR4 signal transduction by binding to LR
[42][155]. These findings offer insight into the mechanism of post-radiotherapy recurrence and metastasis of liver cancer and may be of use in enhancing the therapeutic efficacy of liver cancer stereotactic body radiation therapy. (
Figure 2).
Figure 2. The role of carcinoma-associated fibroblasts (CAFs) and TLR4 signaling in the development and progression of HCC. TLR4 activation is necessary for the development of fibrosis and cancer. CAFs promote tumor growth, angiogenesis, and immunosuppression by producing extracellular matrix components and pro-angiogenic factors. TLR4 signaling is critical in maintaining the immature phenotype of tumor-initiating cells and is implicated in the regulation of fibroblast/CAF-HCC cell interconnection. Created with
Biorender.com.
3. Targeting Toll-like Receptor 4 Signaling to Enhance PD-1 Blockade
3. Targeting TLR4 Signaling to Enhance PD-1 Blockade
Several ICI combinations have shown promising results in the treatment of HCC, including nivolumab (anti-PD-1) plus ipilimumab (anti-CTLA-4), pembrolizumab (anti-PD-1) plus lenvatinib (TKI), which did not manage to outperform lenvatinib in OS and progression-free survival (PFS), and atezolizumab plus bevacizumab, which improved PFS and OS in clinical trials and comprise the mainstay of HCC treatment
[43][156]. Nivolumab plus cabozantinib (TKI), pembrolizumab plus sorafenib (TKI), and atezolizumab plus cabozantinib are currently under clinical trials, while a new approach to strengthening the potential of anti-tumor immunotherapies involves combining immunotherapeutic agents with epigenetic drugs, such as DNMT inhibitors (DNMTi) and HDAC inhibitors (HDACi). The utilization of these inhibitors can lower PD-L1 expression and increase the effectiveness of PD-L1-blocking antibodies against tumors
[44][157].
The pyroptosis signaling pathway, a type of programmed cell death that triggers inflammation, comprises another potential therapeutic target to enhance the immunotherapy response
[45][158]. Gasdermin E in tumor cells increases the infiltration of immune cells into the TME, such as T and NK cells, inducing pyroptosis. Inflammation leads to the release of tumor antigens, which can then activate the immune system’s anti-tumor response
[46][159]. Lv et al. suggested that targeting the protein gasdermin D (GSDMD) in HCC cells can sensitize them to anti-PD-1 therapy, which was achieved by activating the cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS) pathway and downregulating PD-L1 expression
[47][160]. They also provided evidence that targeting GSDMD in HCC cells leads to the release of DNA from dying cells, activating the cGAS pathway
[47][160]. The cGAS pathway is activated by the binding of cytoplasmic DNA to the cGAS enzyme, which produces the second messenger cGAMP (cyclic GMP-AMP). cGAMP then binds to the protein stimulator of interferon genes (STING) which triggers the production of type I IFN. The HMGB1/TLR4/caspase- 1 signaling activated the cGAS pathway, leading to the release of DNA from cells and thus creating a positive feedback of continuous activation
[47][160]. Thus, an effective treatment option for HCC patients with upregulated GSDMD could be a combination therapy with an anti-PD-1 agent, a GSDMD inhibitor, alongside endogenous or exogenous ligands of TLR4
[47][160].
Besides the activation of the TLR4 signaling pathway in DCs driving the generation of an HCC vaccine
[3][125], Lmdd-MPFG has demonstrated additional immune-modifying effects and the potential to exhibit further effective therapeutic outcomes. Xu et al. explored the combination of Lmdd-MPFG with an anti-PD-1, demonstrating that Lmdd-MPFG enhanced the expression of PD-L1 in HCC cells and sensitized local T cells to respond to anti-PD-1. The mechanism involves the activation of the TLR2/MyD88/NF-κB pathway in TAMs by the Lmdd-MPFG vaccine, recruiting p62 to activate the pathway of autophagy, leading to a shift of TAMs from the M2 polarization to an M1 state, altering the cytokine profile to an antitumor one in the TME
[48][161]. This change restored T cell reactivity to the anti-PD-1 blockade, providing a promising new strategy for HCC treatment.