Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by László Csiba and Version 2 by Fanny Huang.
Ischemic stroke (IS), resulting from insufficient blood supply to the brain, is among the leading causes of death and disability worldwide. A potentially severe complication of the disease itself or its treatment aiming to restore optimal blood flow is hemorrhagic transformation (HT) increasing morbidity and mortality.
- ischemic stroke
- hemorrhagic transformation
- clinical risk factors
- pathophysiology
- biomarkers
1. Introduction
Ischemic stroke (IS) is usually caused by arterial occlusion in the central nervous system. It is among the leading causes of death and disability worldwide [1]. Hemorrhagic transformation (HT), that is, extravasation of blood into the ischemic tissue, is a serious complication worsening outcomes and increasing mortality. HT can develop by the natural course of IS or after reperfusion therapy [2]. HT occurs at different rates, varying between 3 and 40% depending on the definition used in different studies [3]. According to the European Cooperative Acute Stroke Study (ECASS), HT on CT scans is divided into two stages: radiologically detectable petechiae can be referred to as hemorrhagic infarction (HI), while more severe forms occur as parenchymal hematoma (PH), with or without mass effect [4]. The National Institutes of Neurological Disorders and Stroke (NINDS) classified HT into asymptomatic and symptomatic forms [5]. The SITS-MOST criteria define symptomatic intracranial hemorrhage (sICH) as a local or remote type 2 parenchymal hematoma occurring within 22 to 36 h post-thrombolysis on a CT scan and is associated with an increase in NIHSS of four points from the baseline or resulting death [6].
2. Pathophysiology of Hemorrhagic Transformation
The integrity of the blood–brain barrier (BBB), i.e., the physiological barrier between the circulating blood and brain interstitium, is crucial. This layer is composed of several cells, including endothelial cells, astrocytes, pericytes, neurons, and extracellular matrix (ECM), forming the so-called neurovascular unit (NVU) [7]. In addition to these cells, basement membranes with tight, adherens, and gap junctions between the cells are also important components of the BBB, which serve as a bidirectional barrier in the transport of different substances and protect brain parenchyma from harmful chemicals. Tight junctions limit the paracellular movement of water, ions, and solutes, and form a barrier to substances with a molecular weight greater than 180 Da. The endothelial cells serve as the first-line defense between the circulating blood and brain parenchyma. They are crucial in the regulation of ion movement and the maintenance of selective molecular permeability and integrity. For this role, endothelial cells have an increased number of mitochondria allowing them to generate the high amount of energy required for the maintenance of proper nutrient support and protection of the brain. Therefore, endothelial cells are very sensitive to an optimal oxygen and glucose supply. BBB endothelial cells are distinguished from peripheral ones by their lack of fenestrations and minimal pinocytotic activity permitting much less transcellular (caveolar) transport compared to the peripheral circulation [7][8][7,8]. The interaction of pericytes with endothelial cells is essential for the stability of the BBB. These cells are surrounded by the ECM, providing support and separation, and regulating intercellular communication. The ECM is composed of structural proteins, like laminin, fibronectin, collagen type-IV, elastin, trombospondin, and proteoglycanes, which are susceptible to enzymatic degradation. Astrocytes are also important components of the BBB. Their endfeet are rich in aquaporin-4 water channels giving them an important role in the regulation of brain water content and electrolyte balance. Early ischemia, via ruining these structures and processes, disrupts the BBB [7][9][7,9]. Components of neuroinflammatory mechanisms are activated involving microglia and astrocyte activation and the secretion of proinflammatory cytokines and growth factors like TNF-α, IL-1β, IL-17, INFγ, VEGF, and matrix metalloproteinases (MMPs) [10]. Neutrophils are recruited to the damaged area; they adhere to the vascular endothelial cells, migrate into the brain parenchyma, and their activation adds to the leakage of the BBB via several ways including the production of matrix metalloproteinase-9 (MMP-9), an enzyme participating in the degradation of the BBB [11][12][11,12]. Lymphocytes infiltrate the brain after neutrophils, releasing different pro-and anti-inflammatory cytokines to regulate the BBB [11]. Monocytes, after transferring into the central nervous system, differentiate into macrophages, promoting the expression of certain factors protecting the BBB and contributing to healing mechanisms [13]. A schematic representation of the NVU and BBB and their disruption due to ischemic damage is shown in Figure 1. Along with the loss of BBB function and the consequent edema formation, the autoregulatory capacity of cerebral vasculature is also weakened. This ability of the cerebral vessels is responsible for the preservation of stable blood flow despite wide variations in systemic blood pressure [14]. Both pericytes, due to their contractile properties, and astrocytes, via their endfeet contacts, contribute to the regulation of capillary blood flow and the maintenance of cerebrovascular autoregulation [7]. In acute stroke, the autoregulatory capacity is of pivotal importance, as it ensures perfusion to the ischemic penumbra and also helps to avoid or moderate reperfusion injury that may happen spontaneously or after thrombolytic and endovascular treatment [14][15][16][14,15,16]. Autoregulatory impairment occurs even in case of minor stroke and may exist ipsilaterally to the affected side but can also be a global phenomenon in both hemispheres [15].
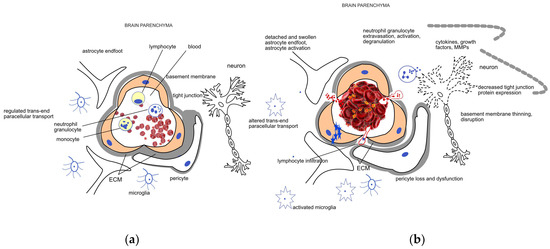
Figure 1. Schematic representation of the (a) healthy neurovascular unit (NVU) and blood–brain barrier (BBB) and (b) their disruption due to acute ischemic damage. The NVU is composed of endothelial cells, pericytes, the extracellular matrix (ECM) surrounding them, astrocytes, and neurons. The main components of the BBB are the endothelial cells, the different junctions between them, and the basement membrane. Endothelial cells, pericytes, and the surrounding ECM ensure a regulated trans- and paracellular transport required for the maintenance of the homeostasis in the brain parenchyma under healthy conditions. Mechanical occlusion of a vessel by a thrombus or an embolus, as shown on the right side of the figure, brings about a cascade of events including endothelial swelling, detachment and dysfunction of pericytes and astrocytes. The trans- and paracellular transport changes (represented by blue stars and red dots, respectively), leading to a sequel of pathophysiological processes including cytotoxic edema. Neutrophil cells are recruited to the damaged area; their extravasation, activation, and degranulation contribute to the neuroinflammatory processes mediated by the activated microglia and several soluble factors. Induction of proteases like matrix metalloproteases (MMPs) further enhance ECM degradation and disruption of the BBB.
Following ischemia, another pathological impact of ischemic stroke is reperfusion occurring spontaneously or after reperfusion therapies. Nevertheless, reperfusion is required for tissue survival; it may also bring on further tissue damage with the potential of HT. Based on experimental data, three phases of restored blood flow may occur, which are presented in Table 1. The initial reperfusional permeability is attributable to an acute elevation of regional cerebral blood flow. This hyperemia, contributing to the acute opening of the endothelial tight junctions, is followed by a period of hypoperfusion with a biphasic permeability response. The hypoperfusion can be attributed to metabolic depletion, microvascular obstruction by cumulating leukocytes, endothelial swelling, and disintegration of the NVU. Hypoperfusion of the ischemic area further results in deficient nutritional support. Enhanced neutrophil adhesion and subsequent inflammatory mechanisms lead to the next period of increased BBB permeability (the first phase of the biphasic permeability 3–6 h after reperfusion). Angiogenesis and increased vasogenic edema are components of the second phase of the biphasic BBB permeability 18–96 h after reperfusion. The duration of the ischemia, degree of reperfusion, and the applied stroke model influence the multiphasic nature of this process. In the ischemic phase, the disturbance of energy production of the cells leads to several pathological processes including failure of the sodium potassium pump and the accumulation of sodium and calcium in the cells causing intracellular translocation of interstitial water resulting in cytotoxic edema. Alternative, less efficient ways of energy production are activated resulting in lactacidosis, directly contributing to the swelling of the cells of the NVU. As a neuronal component, excitotoxic glutamate is released from neurons. Endothelial swelling leads to a shrinking in capillary diameter, further deteriorating blood supply. The induction of proteases like endogenous tissue plasminogen activator and MMPs along with the release of proinflammatory cytokines, chemokines, and adhesion molecules initiate leukocyte infiltration and inflammatory activation. During reperfusion, vasogenic edema develops due to alterations in BBB tight junctions, as an increasing permeability of macromolecules allows fluid movement from intravascular to extravascular spaces. Tight junctions begin to go through a regulated period of disassembly and reassembly between existing endothelial cells, while new assembly is related to new cell growth as angiogenesis starts in the final, biphasic BBB permeability. Vasogenic edema increases overall brain volume with a peak 2–5 days after ischemic stroke; however, neurovascular remodeling may continue weeks after the initial ischemic event [7][9][17][7,9,17].
Table 1. Phases of ischemic and reperfusional damage after acute ischemic stroke.
| Ischemic Phase | Revascularization Phase | |||
|---|---|---|---|---|
| cerebral blood flow | occlusion, decreased cerebral blood flow | acute elevation of cerebral blood flow = hyperemia | decreased blood flow = hypoperfusion | |
| permeability | transcellular increase | initial reperfusional permeability | first phase of biphasic permeability | second phase of biphasic permeability |
| onset | immediately | upon spontaneous opening or thrombolysis or MET (minutes, hours) | ||
. Depending on the severity of the extravasation of blood cells, petechial and parenchymal hemorrhages can be distinguished. Petechial hemorrhage reflects small (HI1) or more confluent (HI2) pinpoint bleeds within the ischemic region. Parenchymal hemorrhage is a more serious form of bleeding involving a larger part of the infarcted area (≤30% in the case of PH1 with a mild space-occupying effect, >30% in the case of PH2 with a significant space-occupying effect) [4].
3. Clinical Risk Factors of Hemorrhagic Transformation
Hypertension, early ischemic signs on CTs, poor collateral circulation, reperfusion therapy, hyperglycemia, severe forms of ischemic stroke (higher NIHSS points), advanced age, low platelet count, and antithrombotic treatment have been known to increase the risk of HT after acute ischemic stroke [17]. Cardioembolic etiology also increases the risk [18]. Table 2 summarizes the clinical risk factors of HT in acute ischemic stroke.
Table 2. Clinical risk factors of HT and their potential resolution.
| Pre-Stroke Risk Factors | Potential Resolution |
|---|---|
| • hypertension | individualized treatment |
| • hyperglycemia | individualized treatment |
| 3–8 (5) h | 18–96 (72) h |
| • advanced age | primary prevention |
| • early ischemic sign | |
| shortening time till reperfusion therapy, potent edema decrease, individualized treatment | |
| • hyperdense middle cerebral artery sign | |
| • calcification of cerebral vessels | |
| • poor collateral circulation | |
| Stroke Factors | |
| • severe form of ischemic stroke | careful patient selection for reperfusion therapy |
| • reperfusion therapy | careful observation, optimal management |
4. Biomarkers of Hemorrhagic Transformation
According to the definition of the World Health Organization (WHO), “any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease” can serve as a biomarker or biological marker [19][81]. Biomarkers are continuously investigated to help with the prediction of potential adverse effects of stroke treatment like HT, optimizing treatment and patient management. The most promising ones are summarized in Table 3.Table 3. Possible biomarkers predicting hemorrhagic transformation after ischemic stroke.
| Biomarker | Abbreviation | Physiological Role | Predictive Value | |
|---|---|---|---|---|
| matrix metalloproteinase 9 | MMP9 | proteolytic enzyme | sensitive and specific marker | |
| cellular fibronectin | c-Fn | major component of the ECM | sensitive and specific marker | |
| ferritin | NA | acute phase protein | neither specific nor sensitive enough | |
| cause | failed intracellular homeostasis, mitochondrial dysfunction | |||
| • liver fibrosis | acute opening of the BBB, loss of cerebral autoregulation | cerebral metabolic depletion, microvascular obstruction, insufficient nutritional support | increased inflammatory activity, angiogenesis | |
| primary prevention, supportive treatment | ||||
| neutrophil-to-lymphocyte ratio | NLR | blood cells | underlying mechanisms | ATP depletion, excitotoxic glutamate efflux from neurons |
| • altered iron homeostasis | use of neuroprotective substances | disassembly of tight junctions | inflammatory and oxidative stress on the BBB, ECM degradation | imperfect tight junction reassembly, new assembly fails to reach the original paracellular impermeability |
| type of edema | cytotoxic | vasogenic | ||
As summarized above, the pathophysiological basis of HT is the disintegration of the BBB and NVUs. Increased BBB leakage promotes extravasation of blood cells initiated by platelets recruiting leukocytes to the sites of their extravasation. The flow of leukocytes decelerates, they start rolling on the activated endothelium, and finally paracellular transmigration happens. This process has been shown to be increased by the acute phase protein fibrinogen in a dose-dependent manner. If the vascular wall is damaged significantly enough, erythrocytes also extravasate after BBB breakdown, resulting in the deposition of hemoglobin-derived neurotoxic products like free iron in the brain parenchyma. The reactive ferrous form of iron triggers lipid peroxidation and oxidative damage resulting in neuronal death and brain edema. Iron-derived reactive oxygen species further alter BBB permeability and promote further transmigration of blood cells. Fibrinogen, apart from its promoting effect on leukocyte extravasation, increases the aggregation of erythrocytes. The source of fibrinogen is the circulation, as it is mainly synthesized by the liver and stored in both the alpha and dense granules of platelets. Upon platelet activation, the deposition of the released fibrinogen serves as a binding site for yet non-activated platelets adding to platelet thrombogenesis and erythrocyte aggregation leading to reduced blood flow and also diminution of the hematoma developing upon HT [7]
| low discriminative ability | ||||
| S100 calcium-binding protein B | ||||
| S100B | glial-specific protein | sensitive but not specific | ||
| • antithrombotic treatment | individualized treatment | |||
| soluble ICAM-1 | sICAM-1 | • low platelet count and mean platelet volume | individualized treatment | |
| adhesive protein involved in inflammatory responses | sensitive but not specific | |||
| vascular adhesion protein-1 | VAP-1 | adhesion molecule | sensitive but not specific | Cranial CT, CTA Signs |
| semicarbazide-sensitive amine oxidase | SSAO | enzyme |