The relevance of genetics in cardiovascular diseases has expanded, especially in the context of cardiomyopathies. Its relevance extends to the management of patients diagnosed with heart failure (HF), given its capacity to provide invaluable insights into the etiology of cardiomyopathies and identify individuals at a heightened risk of poor outcomes. Notably, the identification of an etiological genetic variant necessitates a comprehensive evaluation of the family lineage of the affected patients. In the future, these genetic variants hold potential as therapeutic targets with the capability to modify gene expression.
- heart failure
- genetics
- diagnosis
- prognosis
- therapy
1. Introduction
2. Advancements and Applications of Genetics in Cardiovascular Disease
2.1. Assessment of Pathogenic Genetic Variants
Every individual possesses variants within their genome, ranging from single nucleotide substitutions (single nucleotide variants [SNVs] or single nucleotide polymorphisms [SNPs]) [26,27][7][8] to duplications or deletions spanning entire chromosomes. On average, each person carries about 100 de novo SNVs that emerge during their development [27][8]. Given that protein-coding regions make up roughly 1% of the genome, but contain 85% of disease-causing variants, targeted sequencing frequently focuses on these areas [28][9]. Predominant approaches include sequencing the protein-coding regions of the roughly 20,000 known genes, termed whole-exome sequencing (WES) [29][10] or focusing on a select set of genes associated with a specific clinical condition, often called a “gene panel” (typically focusing on exons only) [30][11]. These techniques can identify minor variants, such as SNVs and minor insertions and deletions. However, the identification of larger and more intricate variants, such as the complete deletion of an exon or complex genomic rearrangements, can be more challenging [31,32][12][13]. The Sanger sequencing technique has been widely used since 1977 for direct sequencing of specific genes. It also serves to validate mutations detected by alternate sequencing approaches and is pivotal in genetic investigations, particularly when a mutated gene is already identified within a family lineage [29,33][10][14]. With the advent of next-generation sequencing (NGS) techniques [34][15] in the early 2000s [35[16][17],36], there has been a shift toward methods that bolster sequencing for increased parallelism and scalability. Presently, high-throughput sequencing stands as the predominant method in many diagnostic contexts [37,38][18][19]. NGS techniques are adept at detecting small variants, including SNVs and minor insertions and deletions. These techniques comprise the following:-
TGP (targeted gene panel): this method examines genes linked to a specific phenotype. However, its scope is primarily confined to selected genes known for their variants, necessitating ongoing updates. The process entails designing a gene panel correlated with a distinct disease and conducting parallel sequencing. It is commonly employed as the primary diagnostic test for probands [31,39][12][20].
-
WES: this method is adept at diagnosing probands presenting with diverse disorders, including pediatric and syndromic cardiomyopathies. It encompasses all genes with the objective of sequencing the complete exome. Through an integrated process, data corresponding to the entire exome can be produced, eliminating the need for additional analyses when updated information becomes available [40,41][21][22].
-
Whole-genome sequencing (WGS): this technique sequences the complete genome, offering diagnostic insights for probands with varied disorders and detailed data on pharmacokinetic variants. While comprehensive, WGS comes with a greater expense and necessitates intricate data analysis. If panel sequencing yields negative results, both WES and WGS stand as viable subsequent diagnostic options [42][23].
2.1.1. Sequencing Modalities
The analytical process begins with the collection of a small sample of peripheral blood or saliva from the patient. At times, samples from parents or close family members may also be required. The DNA essential for sequencing is then extracted from this sample. Upon acquiring the sequences, they undergo analysis and interpretation based on the most current genetic data in the international medical-scientific literature, focusing on the relevant diseases or symptoms. Identified pathological variants are subsequently validated in the original sample using alternative technological methods. In the context of targeted NGS (targeted gene panel, TGP), the assessment of diseases with pronounced genetic heterogeneity and recognized genes is undertaken using designated panels. These panels are of two main types:-
Presequencing panels (targeted resequencing): this approach enables the simultaneous analysis of multiple patients by selectively enriching specific genomic regions prior to sequencing.
- -
-
Cost: The difference in cost is influenced by the reagents used. The use of TGP is more economical if the number of samples per run is optimized. In economic terms, WGS is the most expensive, and although WES is pricier than panels, it can be advantageous depending on the type of study to be performed [46][27].
- -
-
Purpose: Typically, NGS techniques are employed for diseases with high genetic heterogeneity or Mendelian-based genetic diseases (or those suspected to be genetic) where the causative genes remain unidentified. For diseases with established genetic etiology, either custom-designed panels, WES, or WGS can be utilized [45][26].
- -
-
Sensitivity: Sensitivity largely depends on the coverage of the sequences under investigation, that is, the number of reads for specific DNA sections and the overlap extent between these reads. A greater number of reads for a specific region translates to higher sensitivity for that DNA segment [45][26]. During panel analysis, a reduced genome proportion under investigation leads to enhanced coverage and sensitivity. Thus, if a disease is believed to result from a mosaic genomic alteration, NGS panel analysis offers higher sensitivity compared to WES.
- -
-
IFs and VUSs: The probability of identifying IFs and VUSs depends on the genome proportion both analyzed and queried. Analysis based on panels has a reduced association with IFs, since the investigated sequences are directly relevant to the clinical presentation of the proband. In general, a broader sequence analysis correlates with a higher number of IFs and VUSs.
- -
-
Data storage: The extent of the genome analyzed directly influences the volume of data generated. Consequently, suitable platforms for data storage are essential, particularly for analyses yielding substantial data, such as those conducted via WES and, more prominently, WGS [45][26].
2.1.2. Identification and Interpretation of Pathogenic Genetic Variants in Clinical Diagnostics
Various software tools, including Bowtie2, BWA, MAQ, and SOAP2, assist in mapping a patient’s sequences to reference sequences found in databases. Through this method, the patient’s exome is aligned, signifying the alignment phase. After successful alignment, sequence variants can be detected, representing all deviations from the human reference sequence for each gene. This process is known as “variant calling”, an operation that is automated within NGS analysis. Upon detecting approximately 140,000 genetic variants in an individual (a figure representative of the average number derived from WES) through the previously mentioned variant calling operation, it becomes essential to determine their clinical significance. These variants are characterized based on the following:-
Their allele frequency within the general population;
2.2. Variants of Uncertain Significance (VUS)
A mutation denotes a permanent alteration in the nucleotide sequence, whereas a polymorphism represents a variant with a frequency exceeding 1% in the population. Both terms are now more commonly referred to as “variant”, further defined with specific qualifiers such as “pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign”, or “benign”. Some laboratories may incorporate additional levels, particularly for internal use, such as subclassifying VUSs. The designations “likely pathogenic” and “likely benign” imply a confidence level greater than 90% that the variant is, respectively, pathogenic or benign. Importantly, at present, there is no robust data that provides a quantitative assignment of variant certainty to any of the five categories, largely due to the varied nature of many diseases. VUSs are typically overlooked in clinical decision-making, as further information is required to categorize them definitively as either pathogenic or benign. However, the presence of uncertain sarcomere mutations or multiple VUSs in patients with hypertrophic cardiomyopathy (HCM) has been correlated with earlier disease onset and more severe outcomes. Therefore, a deeper understanding of these VUSs becomes imperative for optimized clinical management and better patient outcomes [53][34].CardioBoost uses a disease-specific variant classifier algorithm tailored to predict the pathogenicity of missense variants linked to inherited cardiomyopathies and arrhythmias. One of its distinct advantages is that, when focused on heart disease, this specific variant classifier surpasses the performance of leading whole-genome tools. This superiority underscores the significant potential for improved pathogenicity predictions through disease-specific approaches [55][35]. On the other hand, some tools harness detailed structural information about proteins, examining how mutations might affect protein folding and stability to make their pathogenicity predictions [53][34].
2.3. Classification, Implications, and Clinical Utility of Genetic Variants in Cardiovascular Disease
When a genetic variant correlates with an observed phenotype, laboratories typically categorize it using a five-tier system. If strong evidence supports the variant’s causative role in the condition, it will be labeled as either “pathogenic” or “likely pathogenic”. Conversely, if robust evidence suggests that the variant is not the causative agent for the disease, it will be termed “benign” or “likely benign”. In instances where the evidence is either conflicting or not conclusive, the variant is designated as having “uncertain significance”, often abbreviated as “VUS”. It is crucial to note that VUSs should not be used for cascade testing, and rarely hold clinical significance for the proband [57][36]. Upon identification of a pathogenic or likely pathogenic variant, initiating clinical evaluations and cascade genetic testing for family members is recommended. Family members who test positive for the variant should undergo long-term monitoring, especially if they exhibit the clinical phenotype. In contrast, family members who do not have the variant can generally be exempted from further genetic investigations [22,58][37][38]. In cases where a VUS is identified, supplementary analyses, such as segregation studies and, where feasible, functional studies, should be conducted. Decisions regarding genetic testing and clinical screenings for family members are based on the findings from these analyses. Equally significant, re-evaluation of other potential causes of the disease is warranted when a genetic variant is absent or a genetic test is inconclusive. Clinical screenings of family members should also be considered. PRSs, also known as GRSs, are becoming increasingly relevant in the evaluation of multiple genetic variants [60][39]. These scores consider variants across the genome, each contributing a small amount to disease risk, to compute a cumulative risk score. However, the definitive clinical utility of PRSs in the management of cardiomyopathies remains to be established [61,62,63,64][40][41][42][43]. In the contemporary medical landscape, identifying a genetic variant or assessing an individual’s susceptibility to genetic diseases equates to equipping the patient or their family with a genetic profile (genotype). This profile subsequently stratifies risk considering the identified genetic variant, related diseases, coexisting health conditions, and resultant manifestations (phenotype). Such stratifications can indicate events such as SCD or the onset of clinically evident HF. Furthermore, this genetic “identity card” can dictate whether a patient receives a recommendation for an implantable cardioverter-defibrillator (ICD) insertion, along with all the consequential implications it entails [65][44].2.4. Diagnosis and Counseling
Incorporating genetics into the care regimen for these patients necessitates a structured follow-up and extended surveillance, both for the proband (the initial patient diagnosed with the condition) and their family. This approach not only fosters a more profound comprehension of the disease, but also paves the way for specific and individualized treatments, thereby enhancing both the patient’s prognosis and their clinical management. When diagnosing a patient with cardiomyopathy through genetic testing, the scope extends beyond merely identifying the patient’s present condition. It encompasses continuous genetic assessments for family members and the initiation of a broad counseling framework for both the patient and their family [66,67,68][45][46][47]. For those diagnosed with cardiomyopathy, the genetic trajectory, both for the proband and their relatives, invariably begins with genetic counseling. This step is pivotal, as grappling with an inherited cardiomyopathy can present numerous challenges. Genetic counseling, conducted by healthcare professionals with specific training, strives to assist patients and their families in comprehending and adjusting to the medical, psychosocial, and familial consequences of genetic diseases. It encompasses a range of topics, including understanding inheritance risks, educational sessions on genetic diseases, guidance during genetic testing processes, interpreting genetic variant results, compiling a comprehensive three-generation family medical history, and offering psychosocial support to patients and their families [69][48]. Addressing the communication strategy with family members is a critical component of genetic counseling. The index patient, often referred to as the proband, might choose not to inform at-risk relatives about the inherited condition and the availability of family screening for a variety of reasons [70,71,72][49][50][51]. This situation is concerning, especially given the severe complications and risk of SCD associated with many inherited cardiac conditions. Since many inherited cardiomyopathies exhibit an autosomal dominant inheritance pattern, it is essential to emphasize the importance of both clinical and genetic testing for immediate family members.2.5. Gene Therapy
Gene therapy is a prominent strategy within advanced therapies. At its core, it aims to address the root cause of a disease by providing affected cells with a functional version of a gene. Methods of gene editing include RNA therapy, CRISPR/Cas9 techniques, antisense therapies, and the introduction of innovative drugs [73][52], as summarized in Figure 1. While many clinical phenotypes primarily rely on generic and symptomatic medications, treatments that focus on the underlying cause and offer targeted interventions (specifically, etiology-based and precision therapies) are still in their developmental stages.Research on inherited cardiomyopathies has encountered obstacles due to the lack of suitable in vitro human cardiac cell or tissue models, especially those that replicate patient-specific anomalies. A promising solution is the generation of human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) that are tailored to individual patients [73][52]. Specialized models of hiPSC-CMs, designed to match specific patients or diseases, have been developed, encompassing mutations in genes encoding cardiac sarcomeric and cytoskeletal proteins, ion channels, nuclear proteins, mitochondrial proteins, and lysosomal proteins. The integration of hiPSC-CM technology with genome-editing methods such as the CRISPR/Cas9 system has yielded deeper insights into the genetic origins of diverse cardiomyopathies, leading to the creation of isogenic control lines [74][53]. Transitioning from a cellular perspective to three-dimensional engineered heart tissues, made possible by integrating polymer-based scaffolds with hiPSC-CMs, offers more clinically relevant models for cardiomyopathy studies [75,76][54][55].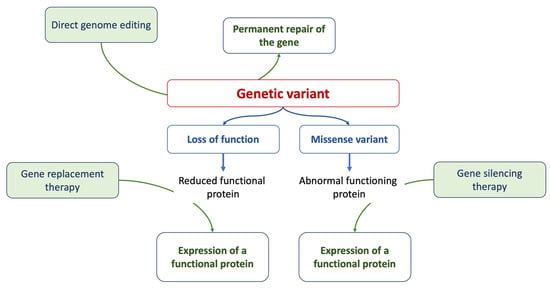 Figure 1.The figure summarizes the current approaches of gene therapy.
Figure 1.The figure summarizes the current approaches of gene therapy.3. Genetics, Cardiomyopathies, and Heart Failure
3.1. Genetic Factors Predisposing to Heart Failure
In patients with cardiomyopathy and those with a genetic variant (i.e., carrier), it is crucial to identify those at an elevated risk of developing HF. The proportion of patients possessing a genetic variant linked to cardiomyopathies who eventually develop HF can differ substantially. This variability can be attributed to multiple factors, including the specific genetic variant in question, its severity, the patient’s family history, age, and other individual risk determinants [100][56]. At present, there are no well-established and scientifically validated multiparametric risk scores that can precisely predict which patients will manifest HF. Nevertheless, a comprehensive evaluation of individual risk should incorporate several parameters. In this context, family history and genetic information can be instrumental in identifying heightened risk. It is well-established that having a first-degree relative with HF augments the risk relative to the broader population [100,109][56][57]. This familial predisposition could potentially stem from shared genetic variants. Various genetic variants have been associated with different types of cardiomyopathies, which can significantly influence the course and outcome of the disease. For example, in DCM, there is evidence that patients with TTN truncation variants might be more susceptible to developing HF when exposed to stressors such as alcohol, pregnancy, or cardiotoxic drugs. Furthermore, those with DCM who carry sarcomeric rare variants or RNA-binding motif protein 20 (RBM20) variants generally experience a more rapid disease progression, sometimes leading to the necessity for interventions such as heart transplantation. Variations in the LMNA gene can have diverse effects on cardiomyocyte structure and function. In the context of HCM, a large multicenter cohort study has shown that a sarcomere mutation is associated with an earlier onset of the disease and serves as a strong predictor of unfavorable clinical outcomes, including the occurrence of VAs and HF [111][58]. The MYH7 gene is a significant genetic factor responsible for encoding the β-myosin heavy-chain (β-MHC) subunit of cardiac myosin. Alterations in this myosin directly influence myocardial mechanical function, contributing to impaired myocardial performance that can result in HF [112][59]. Individuals carrying MYBPC3 missense VUSs demonstrate an elevated incidence of adverse clinical outcomes. These outcomes include VAs, HF, all-cause mortality, atrial fibrillation, and stroke, similar to those with pathogenic MYBPC3 variants [113][60]. Evidence indicates that patients with MYH7 gene mutations present with more severe disease manifestations compared to those with MYBPC3 mutations [114][61]. Moreover, patients harboring MYH7 or MYBPC3 mutations also face a heightened risk of SCD [115][62]. In the context of ARVC, it has been observed that patients with mutations in the DSP, desmocollin-2 (DSC2), and DSG2 genes more commonly exhibit a diminished left ventricular ejection fraction (LVEF) of ≤45% on cardiac magnetic resonance (CMR) imaging in comparison to those with PKP2-related ARVC (27% vs. 4%, p < 0.01) [116][63].3.2. Genetics in Assessing the Risk of Heart Failure Progression
Genetics can be pivotal in guiding the clinical management of patients diagnosed with cardiomyopathy, especially those with established HF. Understanding both the patient’s clinical phenotype and genetic background provides the HF specialist with a unique profile (genotype) for that specific patient, enabling more accurate predictions regarding disease progression and outcomes. Currently, there are no predefined and scientifically validated multiparametric risk scores that can accurately project which patients will develop HF or experience clinical deterioration [100,109][56][57]. Nonetheless, certain genetic variants have been linked to a heightened risk of malignant VAs and progression to advanced HF [4,110][4][64]. Routine clinical evaluations, complemented by echocardiography or CMR every 1–2 years and cardiopulmonary exercise testing every 2–3 years, serve as reliable tools to detect early indicators of disease progression. In this context, timely clinical evaluation and preventive strategies are of paramount importance. Leveraging genetic insights allows for enhanced risk stratification in patients with cardiomyopathy [122][65].3.3. Arrhythmic Risk Stratification
In individuals with cardiomyopathies, atrial fibrillation stands out as the most prevalent arrhythmia across all phenotypes. This condition is linked to an elevated risk of HF, cardioembolic events, and increased mortality. The primary concern for clinicians, however, is managing the risk of VAs leading to SCD, as well as sustained VAs and electrical storms [123][66]. Indeed, while the implantation of an ICD is advocated for secondary prevention, determining its use in patients who have not yet experienced sustained symptomatic VAs remains a complex clinical decision. In DCM, individuals carrying pathogenic variants such as DSP, LMNA, PLN, folliculin (FLCN), TMEM43, and RBM20 demonstrate an increased incidence of notable arrhythmic events [124,125][67][68]. This elevated risk is observed irrespective of their LVEF, and these patients also tend to follow a more challenging clinical trajectory compared to DCM patients not carrying these specific genetic variants. Genes associated with an increased risk of arrhythmias include those encoding nuclear envelope proteins (TMEM43, LMNA, and emerin [EMD]), as well as cytoskeletal and desmosomal proteins (PKP2, DSP, DSC2, and DSG2) [122][65]. Patients with DCM-causing variants in these high-risk genes should be viewed as having a pronounced genetic predisposition to SCD. The decision to implant an ICD for primary prevention in those with LMNA-related cardiomyopathies might be informed by the LMNA-risk ventricular tachyarrhythmia (VTA) calculator, which evaluates the risk of experiencing a life-threatening VTA within a 5-year period [122][65]. This calculator provides a more nuanced risk assessment compared to the prevailing standard of care [4], which identifies high risk based on the presence of ≥2 of the following factors: male gender, non-missense mutations, nonsustained ventricular tachycardia, and an LVEF of ≤45%. In individuals without a high-risk genotype and an LVEF ≥ 35%, the use of LGE on CMR imaging becomes an essential tool for risk assessment [127][69]. It is recommended that genetic testing, encompassing at least the PLN, LMNA, FLNC, and RBM20 genes, be conducted for patients with DCM and atrioventricular (AV) conduction delay before the age of 50, or if a family history of SCD in a first-degree relative is present [122][65]. For DCM patients carrying a pathogenic LMNA gene mutation, considering the placement of an ICD for primary prevention is essential, particularly if the projected 5-year risk of encountering life-threatening VA is ≥10%. This consideration is heightened when the coexistence of nonsustained ventricular tachycardia (NSVT) with an LVEF of ≤50% or an AV conduction delay is observed [122,128][65][70]. MYH7-related dilated cardiomyopathy (MYH7-DCM) is characterized by an early onset, pronounced phenotypic manifestation, limited left ventricular reverse remodeling, and a frequent progression to end-stage HF (ESHF). Complications related to HF are more common than the occasional VAs [129][71]. In HCM, NSVT identified during extended (24/48 h) ambulatory electrocardiogram (ECG) monitoring, particularly in individuals below 30 years of age and those who experience NSVT during physical exertion, is significantly associated with an increased risk of SCD [130][72].
3.4. Implications of Genetic Variants in Clinical Decisions
As previously mentioned, determining the genetic variants within a patient through genetic testing provides a distinctive “identity card”, revealing potential clinical phenotypic traits of the individual. Such characteristics might predispose them to malignant VAs, potential SCD, and the initiation and progression of HF symptoms. ndividuals harboring mutations in genes such as DSP, LMNA, PLN, FLCN, RBM20, and TMEM43 are more susceptible to significant arrhythmic events, irrespective of their LVEF, and tend to follow a more challenging clinical trajectory than those with DCM who are genotype-negative. Specifically, a patient with a mutation in the LMNA gene faces an elevated risk of SCD. As a result, the criteria for ICD implantation for these individuals will be more stringent compared to those without such a mutation, but who are still at high risk for SCD. Consequently, patients in this category should consider ICD implantation as a precaution, even when their LVEF is above 35% [4,122][4][65]. Recognizing one’s genetic profile and inherent predispositions will gain paramount importance in the future, as numerous scientific studies have already highlighted. For instance, in peripartum cardiomyopathy, which was historically viewed as an environmental condition, there is a notable prevalence of truncating variants in genes associated with DCM. Thus, it becomes essential to embrace the idea of individual genetic predispositions. This suggests that specific genetic variants might have a direct correlation with either favorable or unfavorable outcomes. This is even applicable to less prevalent cardiomyopathies, where previously only external or secondary causes were considered as potential etiologies [138][73].4. Heart Failure and Genetics
HF emerges as a multifaceted outcome shaped by the interplay of genetic, acquired, and environmental dimensions. Common genetic variants can precipitate its onset, suggesting that these variants, in conjunction with environmental factors, amplify the susceptibility to HF. Empirical evidence robustly underscores the hereditary dimensions of HF. The Framingham database [139[74][75],140], for instance, revealed a marked 70% increase in the likelihood of HF development in individuals with a family history of the condition. Nonetheless, it is pivotal to recognize that numerous cases are also influenced by lifestyle and external environmental determinants. The trajectory of HF evolution is determined by individual predispositions coupled with the intrinsic factors of accompanying comorbidities. Genetic predispositions can influence susceptibility to the initial event that triggers HF, dictate the progression post-onset, and modulate the efficacy of therapeutic strategies [141][76]. Figure 2 summarizes the complexity of the influence of genetic background observed in HF patients.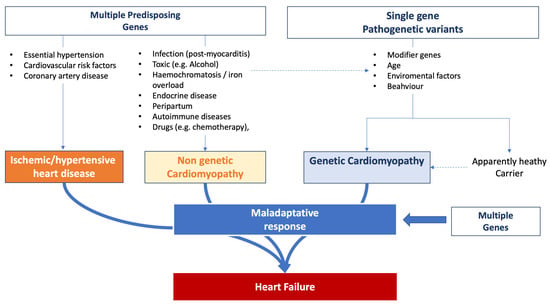 Figure 2.Influence of genetic background on heart failure onset and progression.
Figure 2.Influence of genetic background on heart failure onset and progression.
References
- Shah, R.A.; Asatryan, B.; Sharaf Dabbagh, G.; Aung, N.; Khanji, M.Y.; Lopes, L.R.; Van Duijvenboden, S.; Holmes, A.; Muser, D.; Landstrom, A.P.; et al. Genotype-first approach I. Frequency, penetrance, and variable expressivity of dilated cardiomyopathy-associated putative pathogenic gene variants in UK Biobank participants. Circulation 2022, 146, 110–124.
- De Marvao, A.; McGurk, K.A.; Zheng, S.L.; Thanaj, M.; Bai, W.; Duan, J.; Biffi, C.; Mazzarotto, F.; Statton, B.; Dawes, T.J.W.; et al. Phenotypic expression and outcomes in individuals with rare genetic variants of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 1097–1110.
- McGurk, K.A.; Zheng, S.L.; Henry, A.; Josephs, K.; Edwards, M.; De Marvao, A.; Whiffin, N.; Roberts, A.; Lumbers, T.R.; O’Regan, D.P.; et al. Correspondence on “ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG)” by Miller et al. Genet. Med. 2022, 24, 744–746.
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.N.; et al. Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation 2019, 140, 293–302.
- Van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; Van Der Kooi, A.J.; Van Tintelen, J.P.; Van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500.
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal definition and classification of heart failure: A report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J. Card Fail. 2021, 27, 387–413.
- Conrad, D.F.; Keebler, J.E.; DePristo, M.A.; Lindsay, S.J.; Zhang, Y.; Casals, F.; Idaghdour, Y.; Hartl, C.L.; Torroja, C.; Garimella, K.V.; et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011, 43, 712–714.
- 1000 Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073.
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101.
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189.
- Visser, M.; Dooijes, D.; van der Smagt, J.J.; van der Heijden, J.F.; Doevendans, P.A.; Loh, P.; Asselbergs, F.W.; Hassink, R.J. Next-generation sequencing of a large gene panel in patients initially diagnosed with idiopathic ventricular fibrillation. Heart Rhythm 2017, 14, 1035–1040.
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784.
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376.
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467.
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A prospective study of sudden cardiac death among children and young adults. N. Engl. J. Med. 2016, 374, 2441–2452.
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380.
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59.
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135.
- Michels, V.V.; Moll, P.P.; Miller, F.A.; Tajik, A.J.; Chu, J.S.; Driscoll, D.J.; Burnett, J.C.; Rodeheffer, R.J.; Chesebro, J.H.; Tazelaar, H.D. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1992, 326, 77–82.
- Whiffin, N.; Karczewski, K.J.; Zhang, X.; Chothani, S.; Smith, M.J.; Evans, D.G.; Roberts, A.M.; Quaife, N.M.; Schafer, S.; Rackham, O.; et al. Characterizing the loss-of-function impact of 5’ untranslated region variants in 15,708 individuals. Nat. Commun. 2020, 11, 2523.
- Schwartz, P.J.; Crotti, L.; George, A.L., Jr. Modifier genes for sudden cardiac death. Eur. Heart J. 2018, 39, 3925–3931.
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255.
- Hanchard, N.A.; Umana, L.A.; D’Alessandro, L.; Azamian, M.; Poopola, M.; Morris, S.A.; Fernbach, S.; Lalani, S.R.; Towbin, J.A.; Zender, G.A.; et al. Assessment of large copy number variants in patients with apparently isolated congenital left-sided cardiac lesions reveals clinically relevant genomic events. Am. J. Med. Genet. A 2017, 173, 2176–2188.
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193.
- Giudicessi, J.R.; Lieve, K.V.V.; Rohatgi, R.K.; Koca, F.; Tester, D.J.; van der Werf, C.; Martijn Bos, J.; Wilde, A.A.M.; Ackerman, M.J. Assessment and validation of a phenotype-enhanced variant classification framework to promote or demote RYR2 missense variants of uncertain significance. Circ. Genom. Precis. Med. 2019, 12, e002510.
- Biesecker, L.G.; Green, R.C. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014, 370, 2418–2425.
- Seleman, M.; Hoyos-Bachiloglu, R.; Geha, R.S.; Chou, J. Uses of Next-Generation Sequencing Technologies for the Diagnosis of Primary Immunodeficiencies. Front. Immunol. 2017, 8, 847.
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 15 September 2023).
- Available online: https://www.omim.org (accessed on 15 September 2023).
- Available online: http://genetics.bwh.harvard.edu/pph2/ (accessed on 15 September 2023).
- Available online: https://www.mutationtaster.org (accessed on 15 September 2023).
- Available online: https://www.jcvi.org/research/provean (accessed on 15 September 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424.
- Doh, C.Y.; Kampourakis, T.; Campbell, K.S.; Stelzer, J.E. Basic science methods for the characterization of variants of uncertain significance in hypertrophic cardiomyopathy. Front. Cardiovasc. Med. 2023, 10, 1238515.
- Zhang, X.; Walsh, R.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Govind, R.; Li, N.; Ahmad, M.; Mazzarotto, F.; et al. Disease-specific variant pathogenicity prediction significantly improves variant interpretation in inherited cardiac conditions. Genet. Med. 2021, 23, 69–79.
- Wong, E.K.; Bartels, K.; Hathaway, J.; Burns, C.; Yeates, L.; Semsarian, C.; Krahn, A.D.; Virani, A.; Ingles, J. Perceptions of genetic variant reclassification in patients with inherited cardiac disease. Eur. J. Hum. Genet. 2019, 27, 1134–1142.
- Ingles, J.; McGaughran, J.; Scuffham, P.A.; Atherton, J.; Semsarian, C. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart 2012, 98, 625–630.
- Ingles, J.; Yeates, L.; O’Brien, L.; McGaughran, J.; Scuffham, P.A.; Atherton, J.; Semsarian, C. Genetic testing for inherited heart diseases: Longitudinal impact on health-related quality of life. Genet. Med 2012, 14, 749–752.
- Lambert, S.A.; Gil, L.; Jupp, S.; Ritchie, S.C.; Xu, Y.; Buniello, A.; McMahon, A.; Abraham, G.; Chapman, M.; Parkinson, H.; et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 2021, 53, 420–425.
- Lahrouchi, N.; Tadros, R.; Crotti, L.; Mizusawa, Y.; Postema, P.G.; Beekman, L.; Walsh, R.; Hasegawa, K.; Barc, J.; Ernsting, M.; et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation 2020, 142, 324–338.
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. HCMR Investigators. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142.
- Tadros, R.; Tan, H.L.; ESCAPE-NET Investigators; El Mathari, S.; Kors, J.A.; Postema, P.G.; Lahrouchi, N.; Beekman, L.; Radivojkov-Blagojevic, M.; Amin, A.S.; et al. Predicting cardiac electrical response to sodiumchannel blockade and Brugada syndrome using polygenic risk scores. Eur. Heart J. 2019, 40, 3097–3107.
- Turkowski, K.L.; Dotzler, S.M.; Tester, D.J.; Giudicessi, J.R.; Bos, J.M.; Speziale, A.D.; Vollenweider, J.M.; Ackerman, M.J. Corrected QT interval-polygenic risk score and its contribution to type 1, type 2, and type 3 long-QT syndrome in probands and genotype-positive family members. Circ. Genom. Precis. Med. 2020, 13, e002922.
- Connolly, S.J.; Hallstrom, A.P.; Cappato, R.; Schron, E.B.; Kuck, K.H.; Zipes, D.P.; Greene, H.L.; Boczor, S.; Domanski, M.; Follmann, D.; et al. Meta-analysis of the implantable cardioverter defibrillator secondary prevention trials. Eur. Heart J. 2000, 21, 2071–2078.
- Chivulescu, M.; Lie, Ø.H.; Popescu, B.A.; Skulstad, H.; Edvardsen, T.; Jurcut, R.O.; Haugaa, K.H. High penetrance and similar disease progression in probands and in family members with arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1401–1410.
- Cardim, N.; Freitas, A.; Brito, D. From hypertrophic cardiomyopathy centers to inherited cardiovascular disease centers in Europe. A small or a major step? A position paper from the Nucleus of the Working Group on Myocardial and Pericardial Diseases of the Portuguese Society of Cardiology. Rev. Port. Cardiol. 2011, 30, 829–835.
- Barriales-Villa, R.; Gimeno-Blanes, J.R.; Zorio-Grima, E.; Ripoll-Vera, T.; Evangelista-Masip, A.; Moya-Mitjans, A.; Serratosa-Fernández, L.; Albert-Brotons, D.C.; García-Pinilla, J.M.; García-Pavía, P. Plan of action for inherited cardiovascular diseases: Synthesis of recommendations and action algorithms. Rev. Esp. Cardiol. 2016, 69, 300–309.
- Edwards, A.; Gray, J.; Clarke, A.; Dundon, J.; Elwyn, G.; Gaff, C.; Hood, K.; Iredale, R.; Sivell, S.; Shaw, C.; et al. Interventions to improve risk communication in clinical genetics: Systematic review. Patient Educ. Couns. 2008, 71, 4–25.
- Whyte, S.; Green, A.; McAllister, M.; Shipman, H. Family communication in inherited cardiovascular conditions in Ireland. J. Genet. Couns. 2016, 25, 1317–1326.
- Daly, M.B.; Montgomery, S.; Bingler, R.; Ruth, K. Communicating genetic test results within the family: Is it lost in translation? A survey of relatives in the randomized six-step study. Fam. Cancer 2016, 15, 697–706.
- Kaphingst, K.A.; Blanchard, M.; Milam, L.; Pokharel, M.; Elrick, A.; Goodman, M.S. Relationships between health literacy and genomics-related knowledge, self-efficacy, perceived importance, and communication in a medically underserved population. J. Health Commun. 2016, 21, 58–68.
- de Boer, R.A.; Heymans, S.; Backs, J.; Carrier, L.; Coats, A.J.S.; Dimmeler, S.; Eschenhagen, T.; Filippatos, G.; Gepstein, L.; Hulot, J.S.; et al. Targeted therapies in genetic dilated and hypertrophic cardiomyopathies: From molecular mechanisms to therapeutic targets. A position paper from the Heart Failure Association (HFA) and the Working Group on Myocardial Function of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2022, 24, 406–420.
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844.
- Fomin, A.; Gärtner, A.; Cyganek, L.; Tiburcy, M.; Tuleta, I.; Wellers, L.; Folsche, L.; Hobbach, A.J.; von Frieling-Salewsky, M.; Unger, A.; et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci. Transl. Med. 2021, 13, eabd3079.
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986.
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547.
- Lee, D.S.; Pencina, M.J.; Benjamin, E.J.; Wang, T.J.; Levy, D.; O’Donnell, C.J.; Nam, B.H.; Larson, M.G.; D’Agostino, R.B.; Vasan, R.S. Association of parental heart failure with risk of heart failure in offspring. N. Engl. J. Med. 2006, 355, 138–147.
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules 2019, 9, 878.
- Yousaf, M.; Khan, W.A.; Shahzad, K.; Khan, H.N.; Ali, B.; Hussain, M.; Awan, F.R.; Mustafa, H.; Sheikh, F.N. Genetic Association of Beta-Myosin Heavy-Chain Gene (MYH7) with Cardiac Dysfunction. Genes 2022, 13, 1554.
- Tudurachi, B.S.; Zăvoi, A.; Leonte, A.; Țăpoi, L.; Ureche, C.; Bîrgoan, S.G.; Chiuariu, T.; Anghel, L.; Radu, R.; Sascău, R.A.; et al. An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 10510.
- Velicki, L.; Jakovljevic, D.G.; Preveden, A.; Golubovic, M.; Bjelobrk, M.; Ilic, A.; Stojsic, S.; Barlocco, F.; Tefelmeier, M.; Okwose, N.; et al. Genetic determinants of clinical phenotype in hypertrophic cardiomyopathy. BMC Cardiovasc. Disord. 2020, 20, 516.
- Herrera-Rodríguez, D.L.; Totomoch-Serra, A.; Rosas-Madrigal, S.; Luna-Limón, C.; Marroquín-Ramírez, D.; Carnevale, A.; Rosendo-Gutiérrez, R.; Villarreal-Molina, M.T.; Márquez-Murillo, M.F. Genes frequently associated with sudden death in primary hypertrophic cardiomyopathy. Arch. Cardiol. Mex. 2020, 90, 58–68.
- Christensen, A.H.; Platonov, P.G.; Jensen, H.K.; Chivulescu, M.; Svensson, A.; Dahlberg, P.; Madsen, T.; Frederiksen, T.C.; Heliö, T.; Lie, Ø.H.; et al. Genotype-phenotype correlation in arrhythmogenic right ventricular cardiomyopathy-risk of arrhythmias and heart failure. J. Med. Genet. 2022, 59, 858–864.
- De Backer, J.; Narula, J. Cardiolaminopathies: Weighing in on the Concept of Genotype-First Screening. J. Am. Coll. Cardiol. 2022, 80, 60–62.
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626.
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126.
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307.
- Verstraelen, T.E.; van Lint, F.H.M.; Bosman, L.P.; de Brouwer, R.; Proost, M.V.; Abeln, B.G.S.; Taha, K.; Zwinderman, A.H.; Dickhoff, C.; Oomen, T.; et al. Prediction of ventricular arrhythmia in phospholamban p.Arg14del mutation carriersreaching the frontiers of individual risk prediction. Eur. Heart J. 2021, 42, 2842–2850.
- Mirelis, J.G.; Escobar-Lopez, L.; Ochoa, J.P.; Espinosa, M.Á.; Villacorta, E.; Navarro, M.; Casas, G.; Mora-Ayestarán, N.; Barriales-Villa, R.; Mogollón-Jiménez, M.V.; et al. Combination of late gadolinium enhancement and genotype improves prediction of prognosis in non-ischaemic dilated cardiomyopathy. Eur. J. Heart Fail. 2022, 24, 1183–1196.
- Ader, F.; De Groote, P.; Réant, P.; Rooryck-Thambo, C.; Dupin-Deguine, D.; Rambaud, C.; Khraiche, D.; Perret, C.; Pruny, J.F.; Mathieu-Dramard, M.; et al. FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations. Clin. Genet. 2019, 96, 317–329.
- de Frutos, F.; Ochoa, J.; Navarro-Peñalver, M.; Peñalver, M.; Baas, A.; Bjerre, J.V.; Zorio, E.; Méndez, I.; Lorca, R.; Verdonschot, J.A.J.; et al. Natural History of MYH7-Related Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 80, 1447–1461.
- Gimeno, J.R.; Tomé-Esteban, M.; Lofiego, C.; Hurtado, J.; Pantazis, A.; Mist, B.; Lambiase, P.; McKenna, W.J.; Elliott, P.M. Exercise-induced ventricular arrhythmias and risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2009, 30, 2599–2605.
- Cooper, L.T., Jr.; Čiháková, D. Do Genes Influence Susceptibility to Myocarditis? JACC Basic. Transl. Sci. 2021, 6, 593–594.
- Fox, C.S.; Evans, J.C.; Larson, M.G.; Kannel, W.B.; Levy, D. Temporal trends in coronary heart disease mortality and sudden cardiac death from 1950 to 1999: The Framingham Heart Study. Circulation 2004, 110, 522–527.
- Lloyd-Jones, D.M.; Wilson, P.W.F. Framingham risk score and prediction of lifetime risk for coronary heart disease. Am. J. Cardiol. 2004, 94, 20–24.
- Skrzynia, C.; Berg, J.S.; Willis, M.S.; Jensen, B.C. Genetics and heart failure: A concise guide for the clinician. Curr. Cardiol. Rev. 2015, 11, 10–17.