Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Wiley Braxton Gillam V and Version 2 by Fanny Huang.
Post-Traumatic Stress Disorder (PTSD) is characterized by a wide-ranging set of symptoms including but not limited to intrusive and distressing memories, dissociative reactions, and serious psychological distress in response to stimuli that resemble a previously experienced traumatic event.
- Post-Traumatic Stress Disorder
- traumatic brain injury
- stroke
1. Introduction
PTSD is characterized by a wide-ranging set of symptoms including but not limited to intrusive and distressing memories, dissociative reactions, and serious psychological distress in response to stimuli that resemble a previously experienced traumatic event [1]. Common to all individuals diagnosed with PTSD is exposure to trauma. However, this trauma may be physical or emotional, suggesting that the disorder does not require neurologic injury. Though it may not be inherent in all cases, many studies indicate a correlation between traumatic brain injury (TBI) and PTSD [2][3][4][5][6][2,3,4,5,6]. One meta-analysis found a 13.5% prevalence rate of PTSD in civilians who had been diagnosed with mild traumatic brain injury (mTBI) [2]. Another study found that greater than 17% of United States Army soldiers with mTBI returning from Operation Enduring Freedom (OEF) in Afghanistan and Operation Iraqi Freedom (OIF) in Iraq screened positive for PTSD [4].
2. The Pathophysiology of Post-Traumatic Stress Disorder
The major brain structures which have been proved to play some role in PTSD symptoms include the prefrontal cortex (PFC), hippocampus, amygdala, and other downstream elements such as the locus coeruleus (LC) and hypothalamus [7][8][9][13,14,15]. A critical component of the prefrontal cortex negatively regulates amygdala function to prevent hyperactivity. It has been found that PTSD patients exhibit reduced activity in the medial prefrontal cortex (mPFC) and anterior cingulate cortex (ACC) during presentation of trauma-related and non-related aversive stimuli [10][11][16,17]. Further, they have been observed to have decreased volume in the ventromedial prefrontal cortex (vmPFC) and ACC [10][11][12][13][14][16,17,18,19,20] and even to utilize altered pathways from the ACC to the amygdala [15][21]. All these changes in the PFC suggest that amygdala dysregulation may be involved in the pathophysiology of PTSD by erasure of the conventionally strict regulation of fear responses [7][13].
Noradrenaline is a neuromodulatory neurotransmitter system capable of producing a rapid and coordinated response to acute stress. A meta-analysis compiling data from 1388 articles along with several other studies found statistically significant increased levels of noradrenaline in PTSD patients compared to controls, indicating the likelihood of increased noradrenergic tone in these individuals [16][17][18][19][22,23,24,25]. Of note, one study conducted in 2001 observed concentrations of NA in hourly CSF samples (collected over a 6 h period) to be higher in male combat veterans compared to healthy controls [20][26]. Based on these findings, Pietrzak and colleagues conducted a follow-up study to determine whether a reduction in the Norepinephrine Transporter (NET) could be responsible for the increased noradrenergic tone in PTSD patients [21][27]. The researchers found evidence for a significant reduction in the density of NET labeled in the LC in PTSD patients compared to controls [21][27]. This was a profound discovery that seemingly identified a specific neural correlate of PTSD in combat veterans.
As previously mentioned, changes in the PFC found in PTSD patients have downstream impacts on the amygdala. Specifically, it has been found that the amygdala in those with PTSD is hyperactive during exposure to trauma-linked events [22][28]. A critical function of the amygdala is to stimulate the hippocampus during the formation of new memories related to fear-inducing events. A lack of proper control of the amygdala provides a possible explanation as to why PTSD patients experience intrusive memories from prior trauma. Although the results were similar for both trauma-exposed individuals and PTSD patients, meta-analyses observed statistically significant differences between these two groups and healthy controls [23][29]. Reduced cortisol levels and signaling in PTSD patients could be responsible for stronger sympathetic nervous system activation and extreme lucidity of traumatic memories [24][25][26][27][30,31,32,33]. Notably, Robert Sapolsky claimed that, under stress, the body releases glucocorticoids, resulting in downregulation of glucocorticoid receptors, thereafter, leading to hippocampal atrophy in response to chronic stress [28][34]. Only a decade later, he described the relevance this theory has to PTSD, whereby hyperactivation of this stress system in response to trauma could lead to cell death in the hippocampus and eventually cognitive dysfunction [29][35]. Particularly compelling evidence for hippocampal involvement in PTSD was found by several meta-analyses reporting a significant reduction in hippocampal volume in PTSD patients as compared to both healthy controls and trauma-exposed individuals without PTSD [30][31][36,37].
Norepinephrine and indirect signaling from the limbic system including the hippocampus, mPFC, and amygdala act on the paraventricular nucleus of the hypothalamus to trigger the release of corticotropin-releasing hormone, or CRH [32][38]. The HPA axis which is initiated by CRH release is the single most critical hormonal facilitator of stress response in humans. Rodent models have demonstrated that PTSD-associated behaviors are observed in response to CRH injection [33][39] and that CRH receptor binding in the amygdala can produce fear responses [34][40]. Despite substantial evidence implicating CRH in high-stress states and PTSD symptoms, results from other studies complicate the matter. Notably, CRH release in the bed nucleus of the stria terminalis has an inhibitory effect on neurons in the amygdala, which results in a decreased fear response [35][41]. This clearly contradicts the idea that CRH release necessitates amygdala excitation. It is possible that CRH differentially affects the amygdala according to the pathway it uses, but further evidence is needed to confirm this theory.
Several studies have also looked downstream of CRH to determine whether cortisol is involved in the development of PTSD. It has been found that lower concentrations of hair cortisol in military veterans prior to overseas deployment was positively correlated with PTSD symptoms post deployment [36][42]. Likewise, police academy recruits with reduced increases in cortisol while watching videos of traumatic situations were more likely to demonstrate signs of long-term distress and decreased resilience after four years [37][43]. Together, these findings seem to indicate that lower cortisol concentrations predict PTSD symptoms following exposure to trauma. However, a meta-analysis demonstrated that heart rate was the only biological measure obtained post trauma that could predict PTSD [38][44]. Additionally, even if low cortisol concentrations are associated with PTSD, no threshold cortisol concentration has been identified, leaving little clinical applicability. It is evident that more research is needed to clarify the relationship between the HPA axis and PTSD. Figure 1 is a greatly simplified model that serves to clarify the complex pathophysiology of PTSD, but it should be noted that the relationship between these neurological structures and PTSD is still widely contested.
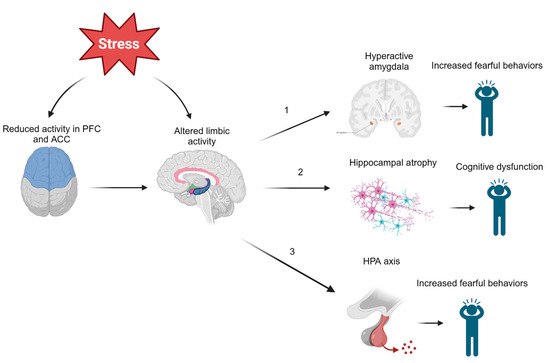
Figure 1. Effects of Modulated Limbic Activity. Chronic or high acute stress experienced during and after trauma may directly and indirectly act on several limbic structures. (1) Altered regulation of the amygdala may induce fearful behaviors observed in those with PTSD. (2) Stress may also lead to hippocampal atrophy, which could be responsible for many symptoms of PTSD, including cognitive dysfunction, involuntary recurrent memories (flashbacks), and more. (3) Limbic structures such as the hippocampus or amygdala may act on the hypothalamus to activate the HPA axis by CRH release. These neuroendocrine disruptions in the body’s primary stress response mechanism could also induce fearful behaviors. Note: ACC = anterior cingulate cortex, PFC = prefrontal cortex, HPA = hypothalamic–pituitary–adrenal axis.