Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Diego Franco and Version 2 by Jessie Wu.
Cancer is a prime example derived from a loss of homeostasis, primarily caused by genetic alterations both in the genomic and epigenetic landscape, which results in deregulation of the gene networks.
- cancer disease
- lncRNAs
1. Introduction
The Cancer Genome Atlas Consortium project, together with other large-scale sequencing projects aimed at characterizing cancer genomes as well as the possible epigenetic and genetic dysregulations that configure them, have provided a precise molecular characterization of approximately 11,000 primary cancers, discovering a substantial fraction of undescribed somatic abnormalities (e.g., point mutations, genetic rearrangements, and copy number alterations) [1][2][3][4][19,20,21,22]. Khuruna et al. (2013) reported that 99% of somatic SNVs in different carcinomas occur in non-coding regions (ncRNAs, pseudogenes, and transcription factor binding sites) [5][23]. Furthermore, a study based on TCGA and lncRNA expression data from TANRIC shows that mutational frequencies in lncRNAs, whose expression is affected by somatic alterations (MutLncs), are low, and that to some extent, their alteration tends to be specific to the type of disease [6][24].
Interestingly, numerous studies revealed a previously uncovered role for long non-coding RNAs (lncRNAs) cRNAs as conditional and constitutive oncogenes or tumor suppressor genes through their facility to regulate each and every characteristic of cancer, such as aberrant proliferation, cellular invasion, altered lipid metabolism, metastasis, and immune escape. Furthermore, regulation exerted by lncRNAs can be carried out both at the transcriptional level - epigenetic and genetic - or at the post-transcriptional level [7][8][9][10][11][12][25,26,27,28,29,30]. These findings, and especially the cancer-specific expression of most of them, pointed to lncRNAs as possible biomarkers or therapeutic targets.
2. Genetic and Epigenetic Contributions of Lolng Non-Coding RNAscRNA Dysregulation in Cancer
The nuclear distribution of lcnRNAs and their interaction with a several nuclear elements, such as transcription factors, chromatin remodeling complexes, or even with a DNA establishing a DNA-RNA complex, have revealed their importance in the regulation of both gene transcription and its epigenetic landscape [13][14][15][31,32,33]. Most types of cancer require numerous changes in nuclear transcription homeostasis in order to escape cellular control systems. These changes translate into upregulation and downregulation of a multitude of genes, oncogenes, and tumor suppressor genes by different mechanisms that result in tumor initiation, progression, and metastasis [16][34]. In line with this, many studies have pointed out lncRNAs as pivotal modulators of nuclear transcription, altering both the transcriptional and epigenetic cellular context responsible for increasing or repressing tumorogenesis as described below (Figure 1).
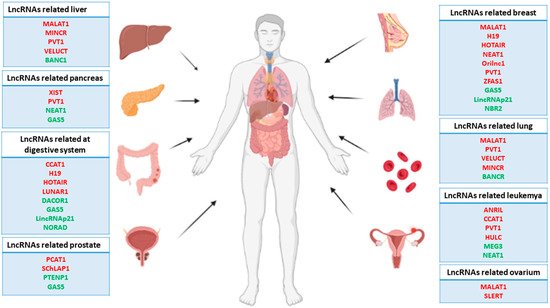
Figure 1. Representative scheme of the main human carcinomas and related lncRNAs. Note that the red lncRNAs act as oncogenes, while the green lncRNAs act as tumor suppressor genes.
Cancer’s underlying epigenetics has been widely studied, given its pivotal role in the initiation, progression, and metastasis of most types of cancer, as well as its potential as a target for different gene therapies [17][18][19][35,36,37]. Epigenetics can be defined as the set of modifications that alters the structure or state of chromatin, differentially regulating its gene expression without changes in the DNA sequence [20][38]. Epigenetic alterations can be included in two basic groups. The first is modifications in the DNA, basically produced by the addition of methyl groups at the 5′ end of certain cytosines located in the so-called CpG islands, where long stretches of CG dinucleotides harbor the promoter regions of the genome that present high, precise, and intense gene regulation [21][39]. Methylation of the cytosines prevents binding of the transcription factors to the promoter regions, which results in gene repression. Interestingly, many types of cancer display a specific pattern of hyper-methylated CpG islands in several tumor suppressor gene promotors, inhibiting their expression and leading to an increase in different subpopulation cancer cells and thus enhanced tumor development [22][23][24][40,41,42]. The second group is modifications in the histone tail, essentially caused by methylation, acetylation, or phosphorylation of certain lysines located in the N-terminal region of histones 3′, altering the nucleosome charge, affecting the chromatin state, and thus allowing or preventing the recruitment of transcriptional co-activators to it [25][43]. These modifications are carried out by chromatin remodeling complexes that add open or close marks to the genome, making it accessible to the transcriptional machinery [26][44]. Unlike the hyper-methylation of the CpG islands, in many types of cancer, hypo-methylation of the genome is observed, leading to ectopic activation of specific oncogenes, whose expression is inhibited in homeostatic conditions. Ectopic activation of these oncogenes is essential for the cell to acquire an oncogenic phenotype and escape from cellular control systems [27][28][45,46]. In the same way, some of these chromatin remodeling complexes show enhanced or reduced activity in malignant cells, which leads to dysregulation in the chromatic structure, promoting the expression of genes that enhance tumor development [29][30][31][47,48,49].
Emerging studies about the role of lncRNAs in carcinogenesis have pointed out as complex regulators in the epigenetics of cancer. As a consequence, many reports of cancer-related lncRNAs have been described as pivotal modulators of the epigenetic landscape by interaction with chromatin at four levels, which are described below: (1) lncRNAs as modulators of histone methylation; (2) lncRNAs as modulators of histone acetylation; (3) lncRNAs as modulators of DNA methylation; (4) lncRNAs as post-transcriptional regulators of the epigenetic apparatus and (5) (Figure 2).
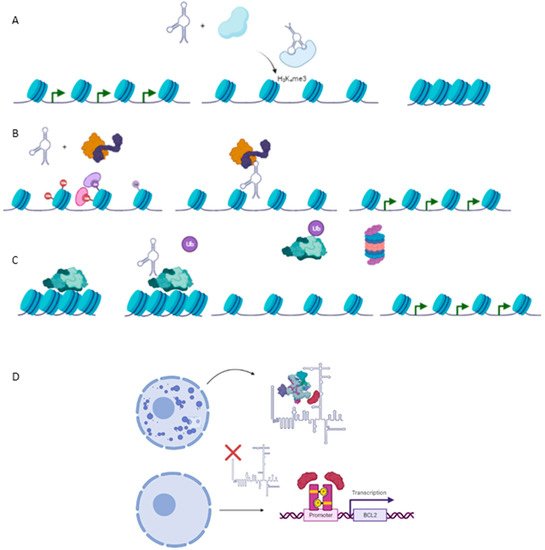
Figure 2. Epigenetic mechanism involved in carcinogenesis. (A) Histone methylation exerted by HOTAIR-PRC2 complex to repress expression of several genes such as Hoxd10, PGR, PCDH, or Jam2 with a protective role against metastasis in breast cancer cell line. (B) TCF21 promoter demethylation exerted by TARID1-GADD45A, which positively modulates expression of the TCF21 gene, a protective factor against head and neck squamous cell carcinoma (HNSCC). (C) Ubiquitination of EZH2, a subunit of PRC2, by ANCR reducing negative marks at Hoxd10 or E-Cadherin genes, exerting a protective role against EMT and metastasis in breast cancer cell line. (D) Required binding SPKQ-Neat1 for the formation of paraspeckles. Downregulation of Neat1 is translated in the high availability of SPKQ and therein the upregulation of apoptosis genes such as BLC2 or BAX.
2.1. Long Non-Coding RNAs as Modulators of Histone Methylation
2.1. lncRNAs as Modulators of Histone Methylation
The interaction between the chromatin remodeling complex PRC2 and HOTAIR was the first reported example of an lncRNA involved in the epigenetic regulation of chromatin [32][50]. In breast cancer, HOTAIR is necessary to recruit PRC2 to its genomic targets, which is required for the latter to establish the repressor marks H3K27me3 in certain tumor suppressor genes that play pivotal roles in the inhibition of metastasis such as Hoxd10, PGR (progesterone receptor), PCDH (Protocadherin 10) or Jam2 (junctional adhesion molecule). Furthermore, a loss of HOTAIR function is sufficient to prevent cell invasion and metastasis in breast cancer, reflecting the role of this lncRNA as a potent oncogene and pointing it out as a possible therapeutic target [33][51].
PRC2 subunits can interact with different nuclear proteins or transcription factors, forming protein complexes that establish epigenetic marks on their genomic targets [34][52]. Tug1, an upregulated lncRNA in different gliomas, acts as a scaffold molecule necessary for PRC2 to interact with YPP1, a neuronal transcription factor. Such a new complex can increase the expression of different genes involved in neuronal differentiation, such as BDNF, NGF, or NFT3, maintaining the pluripotency of malignant cells and thus increasing the aggressiveness of the glioma. Interestingly, the expression of Tug1 is detected in the cytoplasm too, where it acts as a sponge for miR-142, protecting SOX2 and MYC from degradation [35][53].
Maintenance of the epigenetic marks induced by PRC2 requires the participation of another complex: PRC1 [36][54]. ANRIL, a long non-coding RNA located in a genetic desert with a high prevalence of SNPs [37][38][39][55,56,57] (to date, many of these SNPs have been related to a higher prevalence of cancer), interacts with both PRC1 through the CBX7 subunit and PRC2 through the SUZ12 subunit to repress the expression of the CDKN2A/B locus and thus maintaining its silence. This locus is downregulated in many types of cancer, such as leukemia, breast cancer, pancreatic cancer, ovarian cancer, and gliomas [40][41][42][43][44][58,59,60,61,62]. Repression of the CDNK2A/B locus by ANRIL increases the cell proliferation of malignant cells, promoting progression and metastasis. On the contrary, ANRIL depletion reduces cell proliferation and decides the balance toward cell death, therefore reducing the risk of metastasis, pointing to ANRIL as a potent therapeutic target, especially in leukemia and prostate cancer, where it is upregulated [45][46][63,64].
HOTTIP provides another example of lncRNA upregulated in several types of cancer, such as hepatocellular carcinoma, gastric cancer, colorectal cancer, pancreatic cancer, lung cancer, prostate cancer, and osteosarcoma [47][48][49][50][51][52][53][65,66,67,68,69,70,71]. HOTTIP interacts with WDR5, inducing the chromatic opening through the WDR5/MTT complex. This complex upregulates the expression of the HOXA locus through the H3K4me3 marks. In turn, HOXA locus genes repress the expression of several tumor suppressor genes. Additionally, the detection of HOTTIP in the exosome samples of patients has been recognized as a possible prognosis marker in colorectal cancer [54][72].
Unlike the lncRNAs described above, MEG3 represents an intergenetic lncRNA associated with chromatin repressive marks that acts as a potent tumor suppressor gene. MEG3 physically interacts with the EZH2 subunit of the PRC2 complex to repress the expression of several genes involved in TFG-β signaling (e.g., SMAD2 or TGFBR1), increasing the aggressiveness of cancer by promoting invasion and metastasis. This repression is mediated by the formation of a triplex DNA-RNA complex in the GA-rich regions of a gene’s silenced promoters [55][73].
2.2. Long Non-Coding RNAs as Modulators of Histone Acetylation
2.2. lncRNAs as Modulators of Histone Acetylation
Few cases of lncRNAs involved in histone acetylation have been described to date. Wan et al. (2013) described a pivotal role of lncRNA JADE in the earliest steps of DNA damage response (DDR) mechanisms by the modulation of acetylation machinery. As suggested in basic and preclinical studies, DDR is one of the primary anti-cancer barriers during tumorigenesis and is under complex and tight regulation, including alteration of the acetylation patterns of numerous gene promoters [56][57][58][74,75,76]. Clinical studies performed in breast cancer patients have shown lncRNA JADE upregulated expression compared with controls. Mechanistically, lncRNA JADE is required for correct JADE1 expression, a protein necessary to determinate the histone H4 substrate specificity of HBO1, which in turn mediates histone H3–H4 acetylation [59][77]. Several types of cancer display upregulation of HBO1, positively modulating the expression of proliferation-promoting genes linked to a poor cancer prognosis [60][61][78,79]. The depletion of lncRNA JADE is translated in the reduced growth of breast cancer in vivo, as well as in HBO1 deficient cells, suggesting a potential effect as inductors of the proliferation of maligned cells [56][74].
Another case of histone acetylation mediated by lncRNA was reported for lncPRESS1, which plays a pivotal role in the switching of the pluripotent or differentiated state of embryonic stem cells (ESCs) by acting as a decoy molecule from SIRT6. LncPRESS1 competes with SIRT6 for their genomic targets, avoiding that this enzyme can de-acetylate and thus the active gene expression required for promoting cell differentiation [62][80].
2.3. Long Non-Coding RNAs as Modulators of DNA Methylation
2.3. lncRNAs as Modulators of DNA Methylation
The patterns of aberrant methylation of lncRNA promoters have been described in many types of cancer, pointing out their importance in the epigenetic control of carcinogenesis [63][81]. However, only a few cases of lncRNAs involved in DNA methylation have been deeply studied, and their functions remain to be fully elucidated. TARID1 is an intergenic lncRNA whose promoter region is located within the third CpG island of the TFC21 promoter, a known tumor suppressor gene. TARID1 binds GADD45A, a DNA repair protein that promotes active demethylation in numerous promoters. The TARID1-GADD45A complex directs it, together with TDG, a protein necessary for GADD45A to interact with its genomic targets, to the TFC21 promoter, where it eliminates methylation and allows for the expression of TFC21, which in turn plays a key role in protection against head and neck squamous cell carcinoma (HNSCC). Interestingly, TARID1 formed an R-loop with the TCF21 promoter, which was recognized by GADD45A as a region marked for demethylation [64][65][82,83].
Merry et al. (2015) uncovered a total of 148 lncRNAs that are associated with DNMT1 in colon cancer cells through RIP-seq. Among them, one lncRNA was highlighted, named DNMT1-associated colon cancer repressed lncRNA 1 (DACOR1), which is highly and specifically expressed in normal colon tissue while it is repressed in colon cancer cell lines. Furthermore, overexpression of DACOR1 in colon cancer cells resulted in a gain in DNA methylation at multiple loci without changing the DNMT1 expression level. Interestingly, DNTM1 is an important repressor of tumorigenesis [66][84]. ChIRP-seq analysis demonstrated that DACOR1 occupies a total of 338 genomic loci, of which 161 peaks are near 150 annotated genes. Remarkably, 31 of these sites overlapped with differentially methylated regions previously found in colon cancer samples with respect to normal tissues. These findings indicate that DACOR1, cooperating with both chromatin and DNMT1, targets the DNMT1 protein complex toward exact genomic loci. Furthermore, DACOR1 was found to repress the expression of cystathionine β-synthase (CBS) and, in turn, increase methionine, which is the substrate to produce S-adenosyl methionine (SAM). SAM is a necessary methyl donor for DNA methylation in mammalian cells. Thus, DACOR1 may also impinge on DNA methylation through orchestrating cellular SAM levels [67][68][85,86].
2.4. Long Non-Coding RNAs as Post-Transcriptional Regulators of Related Epigenetic Proteins
2.4. lncRNAs as Post-Transcriptional Regulators of Related Epigenetic Proteins
The interaction between lncRNAs and the related epigenetic protein landscape is not limited to recruitment, scaffold, or active element functions required for chromatin remodeling. Furthermore, lncRNAs have been reported as pivotal players in modulating the chromatin protein complex stability by promoting protein degradation, exerting protectives roles in most of cases to trigger pro-oncogenic epigenetic marks or inhibit protein degradation, acting as oncogenes. For example, ANCR is capable of directly binding to the EZH2 subunit, promoting its degradation. ANCR-EZH2 binding is required for CDK1 to target EZH2 for ubiquitin–proteasome degradation via Thr-345 and Thr-487 phosphorylation in breast cancer cells. Curiously, in breast cancer, ANCR expression is inactive, leading to hyperactivity of EZH2, which in turn sets up several repressive marks in tumor suppressor genes such as Hoxa10 or E-Cadherin, which are involved in progression and EMT signals [69][87]. Note that the modulation of ANCR has been probed in other carcinomas [70][71][88,89].
EZH2 degradation is not modulated solely by ANCR. MEG3 promotes EZH2 ubiquitination, leading to upregulation of LATS2, a tumor suppressor kinase that inhibits cell proliferation and metastasis through the Hippo signaling pathway in several types of cancer [72][90]. Gallbladder cancer also displayed a downregulation of MEG3 accompanied by LAST2 downregulation and thus increased cell proliferation and metastasis [73][91].
Unlike ANCR or MEG3, LUCAT1 is considered an oncogene by protecting the protein degradation of DNMT1 in esophageal squamous cell carcinoma. A depletion in LUCAT1 expression is correlated with reduced levels of DNMT1 expression together with the upregulation of UHRF1, a protein involved in ubiquitination and therefore DNTM1 degradation [74][92].
2.5. Long Non-Coding RNAs as Nuclear Environment Modulators
2.5. lncRNAs as Nuclear Environment Modulators
The nuclear compartment not only encloses the chromatin in its different phases and the nucleolus but also contains different structures of mostly irregular shapes, known as nuclear bodies [75][93]. These structures exert pivotal roles in the transcriptional regulation of several pathways related to distinct cellular processes, such as the differentiation, proliferation, or maintenance of homeostasis and, therefore, disease development too [76][77][78][79][94,95,96,97]. Many reports have highlighted the role of these structures in cancer pathogenesis, involving them in the underlying transcriptional regulation of tumorigenesis [80][81][82][98,99,100]. Along different nuclear bodies, paraspeckles, substructures located on the periphery of the nucleus, were first discovered in 2002 [83][101] and have emerged as pivotal regulators of nuclear function modulating. First, they distribute nucleus proteins and are available to interact with chromatin or transcriptional machinery [84][102]. Second, they retain mRNAs, avoiding their transport to the cytoplasm and thereby translation. Interestingly, mRNAs retained by paraspeckles are exported later to the cytoplasm, considerably increasing the number of messenger RNA molecules and their translation. However, the underlying biological processes that determine the export time lapse are poorly understood [85][86][103,104]. Third and finally, they sequester proteins related to miRNA biogenesis and processing [87][105]. The formation of paraspeckles requires the presence of NEAT1, or long non-coding RNA nuclear paraspeckle assembly transcript 1 [88][89][106,107]. NEAT1 is transcribed in two isoforms—Neat1.1 and Neat1.2—displaying different RNA processing, which results in the generation of two different transcripts both in their length and in their structural motifs. While the first isoform is dispensable in paraspeckle biogenesis [90][108], the second isoform is responsible for paraspeckle assembly, constituting a limiting factor for the formation of these nuclear bodies and thus determining the tendency of the nucleus to form them [91][109]. Zeng et al. (2018) reported the pivotal importance of Neat1.2 and therein paraspeckles as promotors of the aggressiveness of Chronic myeloid leukemia (CML). SPKQ, a bivalent protein that can act as a splicing factor required in paraspeckle formation or as a transcription factor exerting the activation of apoptotic proteins such as BLC2, BBC3, or BAX, is associated with Neat1.2 through the C motif. Neat1.2-SPKQ binding reduces the availability of this protein to act as a transcription factor, thus reducing the expression of apoptosis factors BLC2, BBC3, and BAX and leading the cell to escape apoptosis, thereby enhancing the growth and proliferation of CML. Curiously, Neat1.2 expression is downregulated by c-Myc, a known repressor of CML progression. Neat1.2 repression mediated by c-Myc reduces paraspeckle formation and leads to SPKQ binding to the promoters of the apoptotic genes referred to above, activating their expression and consequently promoting apoptosis and achieving a better prognosis [84][102].