1. Cell-Cycle-Specific Mechanisms
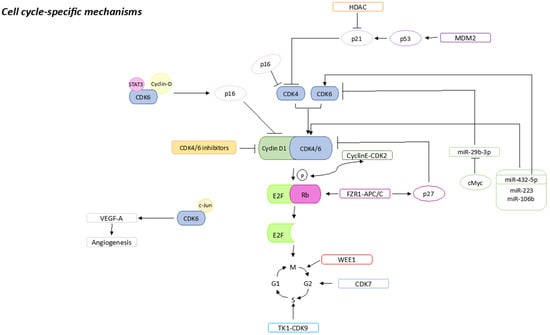
Multiple factors involved in the regulation of the cell cycle are associated with a resistance to CDK4/6is, the loss of drug target genes, and the overexpression of other genes involved in the progression of the cell cycle (Figure 1).
Figure 1.
Cell-cycle-specific mechanisms that could be associated with CDK4/6is resistance. Sharp arrows (→) indicate stimulation, blunt arrows (┴) indicate inhibition.
1.1. pRb Loss or Mutations
pRb is a tumor suppressor protein coded by the RB transcriptional corepressor 1 (RB1) that plays a pivotal role as a cell cycle checkpoint factor. In fact, it is involved in the control of the CDK4/6 pathway, one of the main targets of the CDK4/6is drugs and whose loss, most frequently caused by inactivating mutations of RB1, is the main reason for the resistance to CDK4/6is
[1][2][20,21]. In this case, in spite of this loss, the cell cycle progresses through other molecular pathways, such as the E2F and the cyclin E-CDK2 axis, thus bypassing its dependence on CDK4/6 and causing resistance to CDK4/6is
[3][22]. It has been suggested that administration of cyclin E-CDK2 inhibitors with CDK4/6is may be a valid solution to overcoming resistance
[4][23].
1.2. p16 Amplification
p16 is a tumor suppressor protein belonging to the INK4 family involved in cell cycle regulation if pRb is functional. It is an inhibitor of CDK4, and it has been reported to be an accurate biomarker of pRb loss in different tumors
[5][6][24,25]. p16 amplification is frequently found in BC, and this leads to lower levels of CDK4, thus representing the loss of a target of CDK4/6is. Moreover, p16 amplification is frequently caused by the loss of pRb, thus leading again to resistance to CDK4/6is ex vivo
[7][26]. Palafox et al. suggested that high p16 protein levels and heterozygous RB1 loss-of-function mutations could be predictive in order to identify the CDK4/6is resistance in BC. Moreover, they demonstrated that targeting the p16-CDK4/6 interaction sensitizes p16-overexpressing tumor cells to CDK4/6is
[8][27].
1.3. CDK2 Amplification
Cyclin E is encoded by the CCNE1 gene and its association with CDK2 is involved in cell cycle progression from phase G1 to S. Its main role is the pRb phosphorylation, resulting in complete release of E2F
[9][28]. It has been widely reported that CCNE1 overexpression is responsible for the resistance to CDK4/6is. In fact, cells losing their dependence on CDK4/6 use bypass mechanisms, such as CDK2 amplification, to keep up with cell cycle progression
[10][11][29,30]. Moreover, there is a study that reports that the CDK2 upregulation in ER+ breast cancer is also controlled by TROJAN. TROJAN is a noncoding RNA that can bind to NKRF (NF-Kappa-B-repressing factor) and inhibit its interaction with RELA. This binding leads to CDK2 overexpression and, as aforementioned, leads to CDK4/6is resistance
[12][31].
1.4. E2F Amplification
As a pRb transcription factor, E2F plays an important role in cell cycle regulation. As aforementioned, D-CDK4/6 phosphorylates the pRb leading to E2F release with concurrent transcription of proteins (such as cyclin E) necessary for cell cycle progression. At the same time, cyclin E transcription leads to cyclin E-CDK2 formation, which further phosphorylates pRb. For these reasons, the E2F amplification leads cells to evade CDK4/6 cell cycle regulation thus, driving resistance to their inhibitors
[1][2][9][13][18,20,21,28].
1.5. CDK7 Overexpression
CDK7 regulates the cell cycle by activating the CDK-activating kinase (CAK) and participates in G1 and G2 phases
[14][32]. Martin et al. reported that its overexpression is involved in CDK4/6is resistance
[15][33]. However, details of its involvement in CDK4/6is resistance are not clear, and further studies are needed to better understand its role.
1.6. CDK6 Amplification
The CDK6, together with the CDK4, plays a pivotal role in the cell cycle progression, and its function is mainly kinase-dependent. However, it also upregulates the transcription of p16 in the presence of STAT3 and cyclin D in a kinase-independent way
[16][34]. Moreover, together with c-Jun, CDK6 upregulates the vascular endothelial growth factor A (VEGF-A) which is responsible for cancer progression and drug resistance due to its ability to induce angiogenesis
[17][35]. It was also demonstrated that a particular drug that specifically degrades CDK6 was able to overcome the CDK4/6is resistance
[8][27].
1.7. WEE1 Overexpression
WEE1 is a serine/threonine kinase involved in guaranteeing an accurate DNA replication and, in coordination with CDK1, it is responsible for inhibiting DNA-damaged cells from entering mitosis. Most importantly, it plays a key role in the transition from G2 to M phase of the cell cycle. Its role in the CDK4/6is resistance is not clear; however, it was reported that its inhibition in resistant cells could partially restore the CDK4/6 sensitivity
[18][36].
1.8. MDM2 Overexpression
Mouse double minute 2 homolog (MDM2) is a negative regulator of p53 which is involved in the activation of p21 (a CDK inhibitor), thus leading to cell cycle arrest
[19][20][37,38]. MDM2 overexpression leads to interruption of cell senescence and hence CDK4/6is resistance. For this reason, MDM2 inhibitors may be useful in treating patients’ resistant to CDK4/6is, despite further studies being needed
[21][39].
1.9. HDACs Activation
Histone deacetylases (HDACs) are responsible for removing the acetyl group from the histones’ ε-N-acetyl lysins, playing a crucial role in gene expression regulation
[22][40]. Moreover, they inhibit p21, which interacts with cyclin D
[23][41]. Recently, it has been reported that the CDK4/6i palbociclib resistance may be conferred by HDAC5 depletion through disruption of palbociclib-induced histone deacetylation and suppression of oncogenic gene expression. Thus, the HDAC5 deficiency in cancer cells could be useful to drive the clinical use of CDK4/6is
[24][42].
1.10. FZR1 Loss
Fizzy and the cell division cycle 20-related 1 (FZR1) is a co-activator of the ubiquitin-ligase APC/C that, once activated, can interact with pRb during the cell cycle phase G1
[25][43]. Ruijtenberg et al. showed that FZR1 is a substrate of cyclin D-CDK4/6 and that, once phosphorylated, it loses its ability to activate APC/C. They further demonstrated that knockdown of both FZR1 and APC/C leads cells to bypass their dependence on cyclin D-CDK4/6 for the progression of the cell cycle
[26][44]. The involvement of the APC/C-FZR1 complex in the downregulation of CDK2/4/6 is particularly interesting because it can also upregulate p27 (a CDK inhibitor) through S-phase kinase-associated protein 2 (SKP2) degradation
[27][45]. For all these reasons, the FZR1 loss is linked to resistance to CDK4/6is.
1.11. TK1 Activation
Thymidine kinase 1 (TK1) is an enzyme important for thymidine metabolism during the synthesis of DNA. In resting cells, TK1 activity is low and it increases gradually until it reaches its peak during the S phase. However, it was reported to be continuously overexpressed in patients with different malignancies
[28][46]. It has been demonstrated that TK1 activity is associated with OS and PFS in patients with advanced BC
[29][47]. Moreover, Del Re et al. analyzed exosomal mRNA expression of TK1 and CDK9 in patients with ER+/HER2− mBC enrolled in the ECLIPS biomarker study, showing that higher mRNA expression levels of both of them were linked to palbociclib resistance
[30][48].
1.12. miRNAs Expression
In recent years, microRNA (miRNAs) expression in cancer has been widely studied, due to its important role in cancer progression
[31][49]. Among the different miRNAs identified, some were found to be linked to CDK4/6is resistance, such as miR-432-5p, miR-223, and miR-106b
[32][50]. In particular, miR-432-5p was reported to induce resistance through CDK6 overexpression
[33][51]. Moreover, the oncogene c-Myc could reduce the inhibitory effect of miR-29b-3p on CDK6 by downregulating miR-29b-3p, thus inducing BC resistance to palbociclib
[34][52].
1.13. S6K1 Amplification
Ribosomal protein S6 kinase beta-1 (S6K1) is a serine/threonine protein kinase that acts downstream of the mTORC1 complex and regulates cell size, protein translation and cell proliferation
[35][53]. Recently, it has been reported that S6K1 amplification is responsible for primary resistance to CDK4/6is, mainly linked to c-Myc overexpression that induces hyperactivation of cyclins/CDKs. The authors also suggested the use of mTOR inhibitors combined with CDK4/6is to overcoming resistance
[36][54].
2. Cell-Cycle-Non-Specific Mechanisms
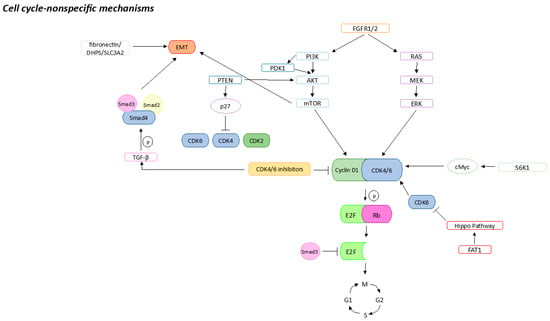
Cell-cycle-non-specific mechanisms include the overexpression of factors that are upstream of the cell cycle, such as FGFR and PI3K/AKT/mTOR, resulting in decreased efficacy of CDK4/6is (Figure 2).
Figure 2.
Cell-cycle-non-specific mechanisms that could be associated with CDK4/6is resistance. Sharp arrows (→) indicate stimulation, blunt arrows (┴) indicate inhibition.
2.1. FGFR Pathway Activation
The fibroblast growth factor receptor 1 (FGFR1) is a tyrosine kinase involved mostly in cell proliferation, differentiation and survival
[37][55]. Mutations in FGFR1 have been widely reported in different cancers, including BC
[38][56]. Different studies report that FGFR1 and FGFR2 are linked to CDK4/6is resistance. In particular, it was reported that this resistance was caused by the amplification of FGFR1, which led to the activation of PI3K/AKT and RAS/MEK/ERK signaling pathways
[39][57]. In addition, a recent study demonstrated that giving a specific FGFR1 tyrosine kinase-inhibitor to resistant cells could revert the resistance
[40][58]. Finally, it was shown that the FGFR2-activating mutation in ER+ BC could contribute to palbociclib resistance and, when the cells were given a high dose of FGFR inhibitors, they could be completely resensitised to the drug.
[41][59]. These studies suggest a potential therapeutic approach to overcoming such resistance.
2.2. PI3K/AKT/mTOR Pathway Activation
Many studies have reported a main role of the PI3K/AKT/mTOR activation in CDK4/6is resistance. In particular, high levels of pyruvate dehydrogenase kinase 1 (PDK1, a protein kinase that acts downstream of PI3K) and activation of the AKT pathway (phospho-S477/T479 AKT) were reported in ribociclib-resistant BC cells. Moreover, inhibition of PDK1 led to higher sensitivity to ribociclib in these cells
[42][60]. Another study revealed that mTORC1/2 may also be involved in CDK4/6is resistance: inhibition of mTOR in ER+ BC results in a decrease in cyclin D1 protein, pRb phosphorylation, and so E2F mediated transcription. In addition, they found that even if cells resistant to CDK4/6is reactivated the CDK-pRb-E2F pathway, they still were sensitive to mTORC1/2 inhibitors
[43][61]. Also, the protein phosphatase and tensin homolog (PTEN) plays a pivotal role in the regulation of the AKT/mTOR pathway. Costa et al. showed that its loss caused a CDK4/6i (ribociclib) resistance by AKT activation. Its loss translated into p27 downregulation which, again, turned into activation of CDK2 and CDK4
[44][62]. Moreover, knockdown of PTEN in CDK4/6is-sensitive cell lines led to the upregulation of CDK6 and resistance to abemaciclib
[45][63]. These data suggest a wide range of drugs that could be used in combination with CDK4/6is in order to prevent/revert resistance.
2.3. MAPK Pathway Activation
The mitogen-activated protein kinase (MAPK) pathway (RAS/RAF/MEK/ERK) is one of the main pathways downstream of FGFR1. It has been reported that selumetinib, a MEK1/2 inhibitor, in association with fulvestrant and palbociclib could inhibit BC proliferation in patients resistant to CDK4/6is
[46][64]. Moreover, it was shown how the overexpression of KRAS, a member of the RAS family, was involved in palbociclib and fulvestrant resistance
[47][65].
2.4. Hippo Pathway Inhibition by FAT1
The Hippo pathway is a pathway reported to have a tumor-suppressive role
[48][66]. FAT atypical cadherin 1 (FAT1) is a cadherin that interacts both with the Hippo pathway and β-catenin, and it is reported to act as a tumor suppressor gene
[49][67]. Interestingly, a study of 348 patients treated with CDK4/6is showed loss of FAT1 in patients who became resistant to CDK4/6is. The resistance was probably caused by the fact that FAT1 loss led to Hippo pathway inhibition, which turned into CDK6 overexpression
[50][68]. However, another study reported that more genetic alterations are needed besides FAT1 loss to have CDK6 overexpression
[45][63]. This suggests that more in-depth studies need to be performed to clarify this pathway and to understand whether its targeting could have a therapeutic effect.
2.5. Epithelial–Mesenchymal Transition
Epithelial–mesenchymal transition (EMT) consists of the transition of cells from an epithelial to a mesenchymal phenotype. EMT is linked with tissue morphogenesis. However, it has been widely reported to be linked also with tissue invasion, drug resistance and metastasis of cancer
[51][69]. It has been reported that inhibition of the CDK4/6 can induce EMT by activating TGF-β-Smad and PI3K/AKT/mTOR pathways. Once activated, TGF-β can phosphorylate and so activate Smad2 and Smad3. These can create a complex with Smad4 and activate EMT transcription factors
[52][70]. In addition, Smad3 inhibition leads to CDK4/6is resistance, probably because it is no longer able to block E2F from the pRb-E2F complex
[53][54][71,72]. Moreover, TGF-β can lead to EMT through activation of the PI3K/AKT/mTOR pathway
[55][73]. Recently, it has also been reported that the fibronectin/DHPS/SLC3A2 signaling axis may be involved in the resistance to CDK4/6is. Galler et al. showed that the targeting of this signaling axis improved palbociclib sensitivity in pRb-negative BC cells
[56][74].
2.6. Apoptosis Failure
The combination of ET with CDK4/6 inhibition has a predominantly cytostatic effect, thus leading to reduced apoptosis
[57][75]. For this reason, it has been suggested that a combination therapy using CDK4/6is and BCL2 inhibitors could inhibit proliferation and induce apoptosis of cancer cells. Moreover, this approach reduced the proliferation of regulatory T cells in the tumor microenvironment
[58][76].
2.7. Stemness Properties
In the last few years, many studies have reported a central role of cancer stem cells (CSCs) in drug resistance. CSCs are a population of cells capable of self-renewal and differentiation potential. Moreover, they show many alterations in different pathways, and for different reasons, and at dates not completely clear, they also play a main role in drug resistance
[59][77]. Interestingly, Wang et al. found that ER+ BC cells treated with palbociclib developed resistance by showing a senescence-like phenotype, which went on to promote stemness. Particularly, PFKFB4 played a major role in this transformation by enhancing glycolysis, and its downregulation resulted in higher palbociclib sensitivity
[60][78]. Thus, even if more studies are needed, targeting CSCs could be a promising therapeutic approach.