Atrial fibrillation (AF) is the most common arrhythmia worldwide and is associated with increased morbidity and mortality. The mechanisms underlying AF are complex and multifactorial. In addition to overall hemodynamic changes due to increased body weight, excess adiposity raises systemic inflammation and oxidative stress, which lead to adverse atrial remodeling. This remodeling includes atrial fibrosis, atrial dilation, decreased electrical conduction between atrial myocytes, and altered ionic currents, making atrial tissue more vulnerable to both the initiation and maintenance of AF. However, much remains to be learned about the mechanistic links between obesity and AF.
- atrial fibrillation
- obesity
- metabolic syndrome
- adiposity
- atrial myopathy
- atrial remodeling
1. Introduction
2. Conceptual Models of AF
There are multiple theories regarding the mechanisms of AF, dating back to the early 1900s. Classical models of AF include rapidly firing atrial ectopic foci, single re-entry circuits, and multiple functional re-entrant circuits [36][7]. Over the past several decades, the multiple re-entrant circuit theory has been the dominant conceptual model of AF. In the re-entry circuit model, there must be (1) triggers to initiate and (2) vulnerable substrates to sustain the circuits [37][8]. AF triggers are mainly located in the pulmonary veins and originate from sleeves of the atrial muscle that extend from the left atrium (LA) several centimeters into the pulmonary veins [38][9]. Ectopic activities that initiate re-entry circuits can be caused either by triggered activity or automaticity. There are two types of triggered activities: early afterdepolarizations (EADs) and delayed afterdepolarizations (DADs). EADs are spontaneous depolarization of the cell membrane during phases 2 and 3 of action potentials and are caused by abnormal openings of calcium channels or sodium channels. DADs are spontaneous depolarization during phase 4 of action potentials and are caused by spontaneous releases of calcium from an internal calcium store into cytosol, which leads to activation of the sodium-calcium exchanger. On the other hand, automaticity is caused by diastolic depolarization from a net inward current during phase 4 of the action potentials, most commonly from the funny current (HCN channels). Unlike cells of the sinoatrial node, which spontaneously initiate action potentials by diastolic depolarization, mature atrial and ventricular myocytes do not express HCN channels and thus lack this capability. In normal atrial tissue, the most common mechanism of ectopic activities that initiates AF is DADs. In animal studies, EADs became important only in the background of existing long-QT syndrome from abnormal sodium channel function [39][10] while automaticity is more likely to contribute to initiation of AF in an end-stage heart failure where current from HCN channels is upregulated as the atrial tissue reverts to a fetal phenotype [39][10].
To understand how vulnerable atrial substrate allows the perpetuation of AF, two proposed models of the re-entrant circuits will be disscussed. One is the “leading circle” hypothesis, where a functional re-entry establishes itself in a circuit the size of the wavelength (WL). Here, WL is a product of conduction velocity (CV), how fast the action potential propagates from cells to cells, and refractory period (RP), the period immediately following stimulation during which cardiomyocytes cannot respond to further stimulation. In this hypothesis, the number of re-entrant circuits is a function of the atrial size and the WL of the re-entry circuits. For example, atrial dilation, a common finding in atria facing chronic volume and/or pressure overload, can support more re-entrant circuits compared to normal-sized atria. Additionally, a shorter WL, either from decreased CV or decreased RP, allows more re-entrant circuits to be sustained in the atria of the same size. Ultimately, having more circuits makes simultaneous termination of all circuits more difficult and thus increases the chance of AF to continue [36][7]. The second model is the “spiral wave” hypothesis, where maintenance of re-entry spirals depends on tissue excitability, propagation strength, RP, and the angle of curvature of the wavefront (the greater wavefront curvature, the slower CV). For instance, lower excitability and slower propagation (e.g., the use of a sodium channel blocker) limit the wavefront curvature, which mandates larger spirals [40,41][11][12].
3. Adipose Tissue Biology
3.1. The Basics of Adipose Tissue
There are three major types of adipose tissue in mammals, each with unique physical and biochemical properties. These are brown, white, and beige adipose tissue. Brown adipose tissue (BAT) accounts for approximately 4% of total body fat. It resides mainly in the supraclavicular, para-aortic, para-vertebral, and supra-renal regions. The main function of BAT is thermogenesis. Therefore, brown adipocytes are small, multilocular, contain a large number of mitochondria, and express uncoupling protein-1, which allows heat generation by uncoupling the mitochondrial electron transport chain, thereby blocking ATP production and dissipating energy in the form of heat [42][13]. White adipose tissue (WAT), on the other hand, is the most abundant type of adipose tissue in the human body. The main function of WAT is storing energy in the form of triglycerides and secreting hormones that regulate hunger/satiety, metabolism, and inflammatory response. White adipocytes are bigger than brown adipocytes. They contain large fat droplets and few mitochondria. WAT can be subdivided into subcutaneous (SAT) and visceral (VAT) adipose tissue. While SAT occupies the space just beneath the layer of the skin, VAT resides deeper and wraps around internal organs, including the heart and blood vessels [42][13]. Compared to SAT, VAT is more vascularized and innervated [43][14]. It is also more metabolically active and plays a major role in energy balance and plasma glucose homeostasis. Adipocytes in VAT express more receptors to glucocorticoids, a key regulatory hormone for serum glucose levels via modifying metabolic adaptations during stress, such as fasting and starvation. VAT is also more sensitive to catecholamine-induced lipolysis due to the increased function of β3-adrenergic receptors [44][15]. The third type of adipose tissue is beige or brown-in-white adipose tissue. Beige adipocytes are more similar to brown adipocytes in that they contain multilocular lipid droplets and numerous mitochondria. However, they reside inside WAT depots. In humans, it is postulated that beige adipocytes are derived from the “browning” of white adipocytes in response to stimulation of β3-adrenergic receptors, for example, during chronic cold exposure or exercise [45][16].3.2. Remodeling of Adipose Tissue during Obesity
In a state of positive energy balance, such as obesity, adipose tissue undergoes several remodeling processes to increase its lipid storage capacity. Adipocytes in WAT undergo hypertrophy (increase in cell size) of existing mature adipocytes and hyperplasia (increase in cell number) of resident adipocyte precursors. Adipocyte hypertrophy that exceeds angiogenesis capacity creates areas of local hypoxia, leading to cell necrosis and subsequently recruitment of macrophages and other immune cells [46][17]. These immune cells adopt proinflammatory phenotypes in an obese state, creating a chronic low-grade systemic inflammatory state leading to a spectrum of metabolic abnormalities, including insulin resistance, type 2 diabetes mellitus, and fatty liver disease [47,48][18][19].4. Mechanisms Underlying Obesity-Related AF
4.1. Hemodynamic Changes
To keep up with the metabolic demand from excess body weight, obese individuals have increased circulating blood volume and thus increased cardiac output via increased stroke volume [15,50][20][21]. Furthermore, the left ventricle (LV) in obese individuals with commonly associated hypertension also faces an additional elevation of an afterload. Chronic increases in wall tension from both increased pressure and chamber dilation lead to LV hypertrophy and LV diastolic dysfunction. Chronically stiff LV and elevated LV filling pressure ultimately cause increased LA filling pressure and LA dilation. This chronic LA stretch leads to a release of atrial natriuretic peptide, angiotensin II, endothelin 1, and transforming growth factor-β, inflammatory cytokines that ultimately lead to atrial myocyte hypertrophy and atrial fibrosis [51][22]. Overall atrial remodeling has a synergistic effect in promoting AF. That is, atrial dilation can support more re-entry circuits while atrial fibrosis causes a reduction of CV and thus WL, which also permits more re-entrant circuits to be sustained in the atrium. In addition to directly altering hemodynamics, obesity also increases the risk of developing AF via obstructive sleep apnea (OSA), a highly prevalent sleep disorder among obese individuals [52][23]. OSA is strongly associated with AF [53][24]. Repetitive forced inspiration against a closed glottis results in a sudden drop in the intrathoracic pressure, which is transmitted to the LA. Repetitive LA stretch may result in remodeling of LA itself and pulmonary vein ostia [54][25], which are the most common sites of AF triggers [38][9]. Additionally, intermittent hypoxemia and hypercapnia can result in sympathetic activation, which increases the frequency of atrial arrhythmia triggering the initiation of AF [55][26].4.2. Roles of Adipokines
Adipokines are paracrines secreted by adipocytes. In addition to controlling homeostasis of serum glucose levels and lipid metabolism, these adipokines also modulate the degree of systemic inflammation and could affect atrial remodeling and thus the risk of AF. Adiponectin is the most abundant adipokine in the body. It has anti-inflammatory and anti-fibrotic effects and is considered “cardioprotective” to vascular endothelial and cardiomyocytes [57][27]. A series of 100 patients undergoing cardiac surgeries demonstrated that increased epicardial fat volume, measured by cardiac computed tomography (CT), was associated with decreased expression of adiponectin in epicardial adipose tissue [58][28], hinting at the role of adiponectin in atrial remodeling and perhaps AF. However, there are mixed findings in terms of the relationship between serum adiponectin and AF. Similarly, current evidence for the role of leptin in AF remains conflicting. In a diet-induced obese mouse model, leptin mediates atrial fibrosis and AF [63][29] and a higher level of serum leptin is associated with obesity [64][30]. However, there are conflicting reports from observational studies on the association between serum leptin levels and AF. For instance, a case-control study of 80 patients with AF showed an increased level of serum leptin compared to the control [61][31] while a sub-analysis of a prospective cohort of 273 patients with type 2 diabetes mellitus showed no association between serum leptin level and AF [60][32]. Apelin, a known cardioprotective adipokine, has been shown in an animal study to reduce angiotensin II-induced atrial fibrosis and atrial fibrillation [65][33]. In humans, a lower serum level of apelin is associated with an increased risk of developing postoperative AF, as observed in a retrospective case-control study of 508 patients who underwent off-pump coronary artery bypass graft (CABG) surgeries [66][34]. Moreover, a lower serum level of apelin is shown to be associated with AF recurrence after an intervention. For example, in a series of 93 patients with persistent AF who underwent cardioversion, individuals with lower levels of apelin at baseline had a higher rate of AF recurrence at the 6-month follow-up [67][35].4.3. Oxidative Stress
In a chronic state of excess energy balance, where the lipid storage capacity of fat depots is exceeded, there is an elevated level of circulating free fatty acids and triglycerides. In this state, cardiomyocytes have an increased uptake of free fatty acids, leading to increased fatty acid β-oxidation [71][36]. Additionally, elevated plasma level of free fatty acids also increases expression of mitochondrial uncoupling protein-3 [72][37], which leads to further increase in mitochondrial fatty acid oxidation and thus increased production of reactive oxygen species (ROS) [73][38]. This overall increase in oxidative stress leads to both structural and electrical remodeling of atrial tissue.4.4. Roles of Epicardial Fat in Obesity-Related AF
As discussed previously, the unique anatomical location of the epicardial fat depot with a shared circulation with the underlying epicardium allows epicardial adipose tissue to exert direct mechanical and chemical influences on underlying atrial tissue. Obese individuals have an increased volume of epicardial fat, as evidenced by an autopsy series dating back to 1933, which showed increased excess of epicardial fat in 136 obese individuals [75][39]. Moreover, an observational study of obese (n = 99) and lean (n = 96) patients with heart failure with preserved ejection fraction showed that epicardial fat thickness measured by transthoracic echocardiogram was 20% thicker in obese patients compared to lean individuals [76][40]. The mechanistic links between epicardial fat and AF are likely multifactorial. In addition to the potential effects of adipokines similar to those secreted from other VAT depots, epicardial adipose tissue poses several other mechanisms due to its unique location relative to atrial myocytes. For example, the proximity of epicardial fat allows direct mechanical compression of the atria [82][41]. Moreover, excess adiposity can lead to the direct infiltration of epicardial adipose tissue into atrial tissue. This is observed in both obesity-induced animal models [83][42] and in humans. In a series of 92 patients undergoing CABG surgeries, analysis of atrial tissue samples demonstrated significant fibro-fatty infiltration in tissue from patients with AF compared to those without AF [83][42].4.5. Atrial Tissue Electro-Anatomical Remodeling in Obesity
Physiologic changes brought on by obesity alter both the electrical and structural properties of atrial tissue at a molecular level, which ultimately create a substrate primed for the initiation and maintenance of AF (Figure 1).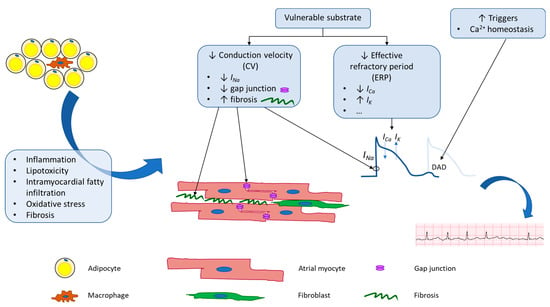
5. Reversibility of Obesity-Induced AF and Remodeling
Multiple studies have demonstrated an association between weight loss and AF burden and progression. An observational study of 1,415 patients with AF who achieved weight loss after participation in a physician-led lifestyle modification program showed that long-term sustained weight loss was associated with maintenance of sinus rhythm [106][58], reduction of AF-associated symptoms [107][59], and delay in progression of AF from paroxysmal to persistent [15,108][20][60]. Another observational study showed that 239 morbidly obese patients who underwent bariatric surgery with resultant weight loss prior to AF catheter ablation had a lower rate of AF recurrence than those who did not undergo bariatric surgery [109][61]. A meta-analysis of patients with weight loss, either by lifestyle modification or bariatric surgery, showed a dose-response reduction in the frequency of AF recurrence after AF ablation [110][62]. Mechanistically, this may be explained by atrial reverse remodeling. In an obese sheep model, weight reduction was associated with decreased atrial fibrosis, increased atrial ERP, improved CV, decreased CV heterogeneity, and decreased AF inducibility [111][63]. As obesity plays a critical role in incident and recurrent AF, the reversibility of this process has significant public health implications as the rate of obesity continues to grow worldwide.6. Conclusions
The global burden of AF is rising with the aging of the population and its associated comorbidities. Obesity-related AF significantly contributes to this burden, as the worldwide prevalence and incidence of obesity in adults and children are also increasing at an alarming rate. Therefore, it is critical to understand the mechanisms underlying obesity-related AF. Not only will this knowledge allow discoveries of novel diagnostic/prognostic tools and more effective treatments, but it will also lead to the development of new strategies for primary and secondary AF prevention. Lastly, it is critical to emphasize the importance of primordial prevention, which includes multidisciplinary effort [130][64] to decrease the worldwide obesity burden.References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528.
- Aliot, E.; Botto, G.L.; Crijns, H.J.; Kirchhof, P. Quality of life in patients with atrial fibrillation: How to assess it and how to improve it. Europace 2014, 16, 787–796.
- Stewart, S.; Murphy, N.F.; Walker, A.; McGuire, A.; McMurray, J.J. Cost of an emerging epidemic: An economic analysis of atrial fibrillation in the UK. Heart 2004, 90, 286–292.
- Kim, M.H.; Johnston, S.S.; Chu, B.C.; Dalal, M.R.; Schulman, K.L. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ. Cardiovasc. Qual. Outcomes 2011, 4, 313–320.
- Lee, W.C.; Lamas, G.A.; Balu, S.; Spalding, J.; Wang, Q.; Pashos, C.L. Direct treatment cost of atrial fibrillation in the elderly American population: A Medicare perspective. J. Med. Econ. 2008, 11, 281–298.
- Nattel, S.; Shiroshita-Takeshita, A.; Brundel, B.J.; Rivard, L. Mechanisms of atrial fibrillation: Lessons from animal models. Prog. Cardiovasc. Dis. 2005, 48, 9–28.
- Bosch, R.F.; Nattel, S. Cellular electrophysiology of atrial fibrillation. Cardiovasc. Res. 2002, 54, 259–269.
- Sanchez-Quintana, D.; Lopez-Minguez, J.R.; Pizarro, G.; Murillo, M.; Cabrera, J.A. Triggers and anatomical substrates in the genesis and perpetuation of atrial fibrillation. Curr. Cardiol. Rev. 2012, 8, 310–326.
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy: A Translational Perspective. Circ. Res. 2020, 127, 51–72.
- Comtois, P.; Kneller, J.; Nattel, S. Of circles and spirals: Bridging the gap between the leading circle and spiral wave concepts of cardiac reentry. Europace 2005, 7 (Suppl. S2), 10–20.
- Pandit, S.V.; Jalife, J. Rotors and the dynamics of cardiac fibrillation. Circ. Res. 2013, 112, 849–862.
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22.
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18.
- Arner, P. Catecholamine-induced lipolysis in obesity. Int. J. Obes. Relat. Metab. Disord. 1999, 23 (Suppl. S1), 10–13.
- Mulya, A.; Kirwan, J.P. Brown and Beige Adipose Tissue: Therapy for Obesity and Its Comorbidities? Endocrinol. Metab. Clin. N. Am. 2016, 45, 605–621.
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101.
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830.
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808.
- Lopez-Jimenez, F.; Almahmeed, W.; Bays, H.; Cuevas, A.; Di Angelantonio, E.; le Roux, C.W.; Sattar, N.; Sun, M.C.; Wittert, G.; Pinto, F.J.; et al. Obesity and cardiovascular disease: Mechanistic insights and management strategies. A joint position paper by the World Heart Federation and World Obesity Federation. Eur. J. Prev. Cardiol. 2022, 29, 2218–2237.
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 968–976.
- Chen, Y.C.; Voskoboinik, A.; Gerche, A.; Marwick, T.H.; McMullen, J.R. Prevention of Pathological Atrial Remodeling and Atrial Fibrillation: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 2846–2864.
- Garvey, J.F.; Pengo, M.F.; Drakatos, P.; Kent, B.D. Epidemiological aspects of obstructive sleep apnea. J. Thorac. Dis. 2015, 7, 920–929.
- Gami, A.S.; Pressman, G.; Caples, S.M.; Kanagala, R.; Gard, J.J.; Davison, D.E.; Malouf, J.F.; Ammash, N.M.; Friedman, P.A.; Somers, V.K. Association of atrial fibrillation and obstructive sleep apnea. Circulation 2004, 110, 364–367.
- Latina, J.M.; Estes, N.A., 3rd; Garlitski, A.C. The Relationship between Obstructive Sleep Apnea and Atrial Fibrillation: A Complex Interplay. Pulm. Med. 2013, 2013, 621736.
- Tavares, L.; Lador, A.; Valderrabano, M. Sleep Apnea and Atrial Fibrillation: Role of the Cardiac Autonomic Nervous System. Methodist DeBakey Cardiovasc. J. 2021, 17, 49–52.
- Villarreal-Molina, M.T.; Antuna-Puente, B. Adiponectin: Anti-inflammatory and cardioprotective effects. Biochimie 2012, 94, 2143–2149.
- Shimabukuro, M.; Hirata, Y.; Tabata, M.; Dagvasumberel, M.; Sato, H.; Kurobe, H.; Fukuda, D.; Soeki, T.; Kitagawa, T.; Takanashi, S.; et al. Epicardial adipose tissue volume and adipocytokine imbalance are strongly linked to human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1077–1084.
- Fukui, A.; Ikebe-Ebata, Y.; Kondo, H.; Saito, S.; Aoki, K.; Fukunaga, N.; Shinohara, T.; Masaki, T.; Teshima, Y.; Takahashi, N. Hyperleptinemia Exacerbates High-Fat Diet-Mediated Atrial Fibrosis and Fibrillation. J. Cardiovasc. Electrophysiol. 2017, 28, 702–710.
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Rev. 2007, 8, 21–34.
- Anaszewicz, M.; Wawrzenczyk, A.; Czerniak, B.; Banas, W.; Socha, E.; Lis, K.; Zbikowska-Gotz, M.; Bartuzi, Z.; Budzynski, J. Leptin, adiponectin, tumor necrosis factor alpha, and irisin concentrations as factors linking obesity with the risk of atrial fibrillation among inpatients with cardiovascular diseases. Kardiol. Pol. 2019, 77, 1055–1061.
- Peller, M.; Kaplon-Cieslicka, A.; Rosiak, M.; Tyminska, A.; Ozieranski, K.; Eyileten, C.; Kondracka, A.; Mirowska-Guzel, D.; Opolski, G.; Postula, M.; et al. Are adipokines associated with atrial fibrillation in type 2 diabetes? Endokrynol. Pol. 2020, 71, 34–41.
- Lv, W.; Zhang, L.; Cheng, X.; Wang, H.; Qin, W.; Zhou, X.; Tang, B. Apelin Inhibits Angiotensin II-Induced Atrial Fibrosis and Atrial Fibrillation via TGF-beta1/Smad2/alpha-SMA Pathway. Front. Physiol. 2020, 11, 583570.
- Xu, S.; Zhang, J.; Xu, Y.L.; Wu, H.B.; Xue, X.D.; Wang, H.S. Relationship between Angiotensin Converting Enzyme, Apelin, and New-Onset Atrial Fibrillation after Off-Pump Coronary Artery Bypass Grafting. BioMed Res. Int. 2017, 2017, 7951793.
- Falcone, C.; Buzzi, M.P.; D’Angelo, A.; Schirinzi, S.; Falcone, R.; Rordorf, R.; Capettini, A.C.; Landolina, M.; Storti, C.; Pelissero, G. Apelin plasma levels predict arrhythmia recurrence in patients with persistent atrial fibrillation. Int. J. Immunopathol. Pharmacol. 2010, 23, 917–925.
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258.
- Murray, A.J.; Panagia, M.; Hauton, D.; Gibbons, G.F.; Clarke, K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes 2005, 54, 3496–3502.
- Cole, M.A.; Murray, A.J.; Cochlin, L.E.; Heather, L.C.; McAleese, S.; Knight, N.S.; Sutton, E.; Jamil, A.A.; Parassol, N.; Clarke, K. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res. Cardiol. 2011, 106, 447–457.
- Smith, H.L.; Willius, F.A. Adiposity of the heart: A clinical study of one hundred and thirty six obese patients. Ann. Intern. Med. 1933, 52, 911–931.
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure with Preserved Ejection Fraction. Circulation 2017, 136, 6–19.
- Wong, C.X.; Ganesan, A.N.; Selvanayagam, J.B. Epicardial fat and atrial fibrillation: Current evidence, potential mechanisms, clinical implications, and future directions. Eur. Heart J. 2017, 38, 1294–1302.
- Haemers, P.; Hamdi, H.; Guedj, K.; Suffee, N.; Farahmand, P.; Popovic, N.; Claus, P.; LePrince, P.; Nicoletti, A.; Jalife, J.; et al. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur. Heart J. 2017, 38, 53–61.
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717.
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 120.
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427.
- Fang, G.; Cao, W.; Chen, L.; Song, S.; Li, Y.; Yuan, J.; Fei, Y.; Ge, Z.; Chen, Y.; Zhou, L.; et al. Cadherin-11 deficiency mitigates high-fat diet-induced inflammatory atrial remodeling and vulnerability to atrial fibrillation. J. Cell. Physiol. 2021, 236, 5725–5741.
- Scott, L., Jr.; Fender, A.C.; Saljic, A.; Li, L.; Chen, X.; Wang, X.; Linz, D.; Lang, J.; Hohl, M.; Twomey, D.; et al. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc. Res. 2021, 117, 1746–1759.
- Otsuka, N.; Okumura, Y.; Arai, M.; Kurokawa, S.; Nagashima, K.; Watanabe, R.; Wakamatsu, Y.; Yagyu, S.; Ohkubo, K.; Nakai, T.; et al. Effect of obesity and epicardial fat/fatty infiltration on electrical and structural remodeling associated with atrial fibrillation in a novel canine model of obesity and atrial fibrillation: A comparative study. J. Cardiovasc. Electrophysiol. 2021, 32, 889–899.
- Okumura, Y.; Watanabe, I.; Nagashima, K.; Sonoda, K.; Sasaki, N.; Kogawa, R.; Takahashi, K.; Iso, K.; Ohkubo, K.; Nakai, T.; et al. Effects of a high-fat diet on the electrical properties of porcine atria. J. Arrhythm. 2015, 31, 352–358.
- Abed, H.S.; Samuel, C.S.; Lau, D.H.; Kelly, D.J.; Royce, S.G.; Alasady, M.; Mahajan, R.; Kuklik, P.; Zhang, Y.; Brooks, A.G.; et al. Obesity results in progressive atrial structural and electrical remodeling: Implications for atrial fibrillation. Heart Rhythm. 2013, 10, 90–100.
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Manavis, J.; Wood, J.P.; Finnie, J.W.; Samuel, C.S.; Royce, S.G.; Twomey, D.J.; et al. Electrophysiological, Electroanatomical, and Structural Remodeling of the Atria as Consequences of Sustained Obesity. J Am. Coll. Cardiol. 2015, 66, 1–11.
- Hohl, M.; Lau, D.H.; Muller, A.; Elliott, A.D.; Linz, B.; Mahajan, R.; Hendriks, J.M.L.; Bohm, M.; Schotten, U.; Sanders, P.; et al. Concomitant Obesity and Metabolic Syndrome Add to the Atrial Arrhythmogenic Phenotype in Male Hypertensive Rats. J. Am. Heart Assoc. 2017, 6, e006717.
- Schram-Serban, C.; Heida, A.; Roos-Serote, M.C.; Knops, P.; Kik, C.; Brundel, B.; Bogers, A.; de Groot, N.M.S. Heterogeneity in Conduction Underlies Obesity-Related Atrial Fibrillation Vulnerability. Circ. Arrhythm. Electrophysiol. 2020, 13, e008161.
- Munger, T.M.; Dong, Y.X.; Masaki, M.; Oh, J.K.; Mankad, S.V.; Borlaug, B.A.; Asirvatham, S.J.; Shen, W.K.; Lee, H.C.; Bielinski, S.J.; et al. Electrophysiological and hemodynamic characteristics associated with obesity in patients with atrial fibrillation. J. Am. Coll. Cardiol. 2012, 60, 851–860.
- McCauley, M.D.; Hong, L.; Sridhar, A.; Menon, A.; Perike, S.; Zhang, M.; da Silva, I.B.; Yan, J.; Bonini, M.G.; Ai, X.; et al. Ion Channel and Structural Remodeling in Obesity-Mediated Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2020, 13, e008296.
- Martinez-Mateu, L.; Saiz, J.; Aromolaran, A.S. Differential Modulation of I(K) and I(Ca,L) Channels in High-Fat Diet-Induced Obese Guinea Pig Atria. Front. Physiol. 2019, 10, 1212.
- Zhang, F.; Hartnett, S.; Sample, A.; Schnack, S.; Li, Y. High fat diet induced alterations of atrial electrical activities in mice. Am. J. Cardiovasc. Dis. 2016, 6, 1–9.
- Pathak, R.K.; Middeldorp, M.E.; Meredith, M.; Mehta, A.B.; Mahajan, R.; Wong, C.X.; Twomey, D.; Elliott, A.D.; Kalman, J.M.; Abhayaratna, W.P.; et al. Long-Term Effect of Goal-Directed Weight Management in an Atrial Fibrillation Cohort: A Long-Term Follow-Up Study (LEGACY). J. Am. Coll. Cardiol. 2015, 65, 2159–2169.
- Abed, H.S.; Wittert, G.A.; Leong, D.P.; Shirazi, M.G.; Bahrami, B.; Middeldorp, M.E.; Lorimer, M.F.; Lau, D.H.; Antic, N.A.; Brooks, A.G.; et al. Effect of weight reduction and cardiometabolic risk factor management on symptom burden and severity in patients with atrial fibrillation: A randomized clinical trial. JAMA 2013, 310, 2050–2060.
- Middeldorp, M.E.; Pathak, R.K.; Meredith, M.; Mehta, A.B.; Elliott, A.D.; Mahajan, R.; Twomey, D.; Gallagher, C.; Hendriks, J.M.L.; Linz, D.; et al. PREVEntion and regReSsive Effect of weight-loss and risk factor modification on Atrial Fibrillation: The REVERSE-AF study. Europace 2018, 20, 1929–1935.
- Donnellan, E.; Wazni, O.M.; Kanj, M.; Baranowski, B.; Cremer, P.; Harb, S.; McCarthy, C.P.; McEvoy, J.W.; Elshazly, M.B.; Aagaard, P.; et al. Association between pre-ablation bariatric surgery and atrial fibrillation recurrence in morbidly obese patients undergoing atrial fibrillation ablation. Europace 2019, 21, 1476–1483.
- Zhao, H.; Li, X.; Yu, P.; Liu, M.; Ma, J.; Wang, J.; Zhu, W.; Liu, X. Association between weight loss and outcomes in patients undergoing atrial fibrillation ablation: A systematic review and dose-response meta-analysis. Nutr. Metab. 2023, 20, 5.
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Wood, J.P.M.; Manavis, J.; Samuel, C.S.; Patel, K.P.; Finnie, J.W.; Alasady, M.; et al. Atrial Fibrillation and Obesity: Reverse Remodeling of Atrial Substrate with Weight Reduction. JACC Clin. Electrophysiol. 2021, 7, 630–641.
- Perone, F.; Pingitore, A.; Conte, E.; Halasz, G.; Ambrosetti, M.; Peruzzi, M.; Cavarretta, E. Obesity and Cardiovascular Risk: Systematic Intervention Is the Key for Prevention. Healthcare 2023, 11, 902.