Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xingkang Wu and Version 2 by Rita Xu.
Psoriasis is an incurable skin disease that develops in about two-thirds of patients before the age of 40 and requires lifelong treatment; its pathological mechanisms have not been fully elucidated. The core pathological process of psoriasis is epidermal thickening caused by the excessive proliferation of epidermal keratinocytes, which is similar to the key feature of cancer; the malignant proliferation of cancer cells causes tumor enlargement, suggesting that there is a certain degree of commonality between psoriasis and cancer.
- psoriasis
- cancer
- cell proliferation
1. Introduction
Psoriasis is one of the most common chronic skin diseases, with clinical features of skin lesions that have raised, well-demarcated, erythematous oval plaques with adherent silvery scales [1]. In addition to skin lesion-related symptoms, psoriatic patients suffer from comorbidities, such as psoriatic arthritis, metabolic syndrome, psychological disorders, cardiovascular disease, atherosclerosis, inflammatory bowel disease, chronic obstructive pulmonary disease, etc. [2]. Psoriasis predominantly develops in young adults, with about two-thirds of patients developing the disease before the age of 40 years, about one-third of patients developing the disease in childhood (age < 18 years), and about 57% of patients suffering from moderate to severe psoriasis [3][4][3,4]. In brief, psoriasis is a disease that seriously endangers human health.
Psoriasis is not yet curable and requires lifelong treatment. The choice of treatment for psoriasis depends on several factors, including location, the severity of the disease, and whether it is accompanied by other diseases [5]. Topical therapy involving corticosteroids, vitamin D analogues, tapinarof, and calcineurin inhibitors is the gold standard for patients with mild psoriasis [6]. Oral systemic therapies are the conventional treatment options for patients with moderate to severe psoriasis, with oral systemic agents such as methotrexate, acitretin, ciclosporin, dimethyl fumarate, apremilast, and tofacitinib [7]. Biological therapies are increasingly used to treat moderate to severe, refractory, and special types of psoriasis [8]. Currently, biologics targeting TNF-α, IL-12/IL-23 p40, IL-23p19, IL-17A, and IL-36R are used in the clinical treatment of psoriasis, and they are superior in terms of efficacy than nonbiological treatment [9][10][9,10]. Phototherapy can be safely combined with other therapeutic options to improve the effects of therapy [11]. Oral small-molecule targeted drugs are promising options for treating psoriasis, providing advantages over biologics in terms of patient convenience, reduced healthcare costs, and improved quality of life [12]. Currently, the available oral small-molecule targeted drugs for treating psoriasis include the phosphodiesterase 4 (PDE4) inhibitor apremilast and the tyrosine-protein kinase 2 (TYK2) inhibitor deucravacitinib [12]. In summary, a range of treatment methods for psoriasis has been developed over the past few decades. Despite the existence of many therapeutical options, psoriasis still cannot be completely cured owing to treatment failure and disease relapse. Therefore, there is an urgent need to investigate the molecular and genetic mechanisms of psoriasis pathogenesis from different perspectives in order to develop new therapeutic targets and drugs.
The pathological mechanisms of psoriasis have not been fully elucidated. The key pathological features of psoriasis are excessive keratinocyte proliferation, abnormal differentiation, and the infiltration of a variety of immunoinflammatory cells. Additionally, its accepted pathogenesis is that the interaction between hyperproliferative epidermal keratinocytes and activated immune cells interact to form a positive feedback loop (Figure 1) [5][13][5,13]. The pathological changes related to psoriasis are similar to the key feature of cancer—that is, the malignant proliferation of cancer cells causes tumor enlargement, suggesting that there is a certain degree of commonality between psoriasis and cancer [13][14][13,14].
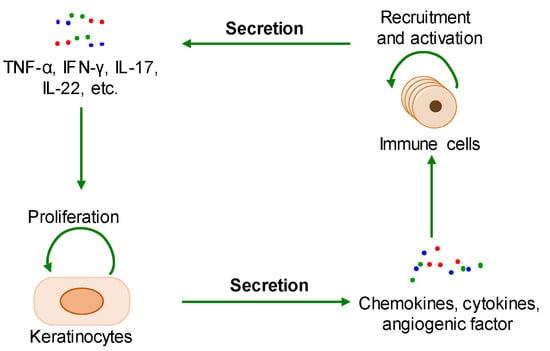
Figure 1. The positive feedback loop of a keratinocyte-immune microenvironment. TNF-α: tumor necrosis factor alpha; INF-γ: interferon-gamma; IL-17: interleukin-17; IL-22: interleukin-22.
2. Common Pathological Mechanisms between Psoriasis and Cancer
A great deal of research has shown that the interaction between cell proliferation and abnormal immune microenvironment, metabolic reprogramming, and epigenetic reprogramming are common pathological mechanisms between psoriasis and cancer, and there are common pathogenic genes or proteins in these shared pathological mechanisms.
2.1. The Interaction between Cell Proliferation and Abnormal Immune Microenvironment
During the onset of psoriasis, external stimuli trigger the recruitment and activation of immune cells to secrete cytokines such as IL-17, IL-22, TNF-α, and INF-γ, which promote keratinocyte proliferation. The proliferative keratinocytes secrete cytokines, chemokines, and angiogenic factors such as TNF-α, IL-8, CXCL8, and VEGF, which promote angiogenesis and the recruitment and activation of immune cells. Thus, the immune microenvironment of psoriatic lesions is characterized by angiogenesis, increasing proinflammatory factors, and immune cells. Additionally, external stimuli also induce keratinocyte proliferation and the secretion of proinflammatory factors [15][16][15,16]. In summary, the proliferative keratinocytes and activated immune cells form a positive loop mediated by proinflammatory factors, maintaining and accelerating the process of psoriasis (Figure 2).
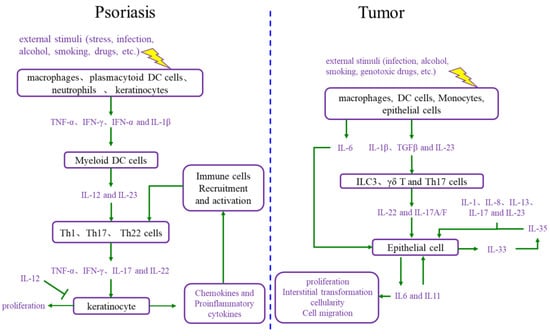
Figure 2. Comparison of the interaction between cell proliferation and immune microenvironment in psoriasis and cancer. DC cells: dendritic cells; IL-12: interleukin-12; IL-23: interleukin-23; Th1 cells: T helper 1 cells; Th17 cells: T helper 17 cells; Th22 cells: T helper 22 cells; TNF-α: tumor necrosis factor alpha; INF-γ: interferon gamma; IL-17: interleukin-17; IL-6: interleukin-6; IL-1β: interleukin-1β; TGFβ: transforming growth factor-β; ILC3 cells: group 3 innate lymphoid cells; γδ T cells: gamma delta T cells; IL-1: interleukin-1; IL-8: interleukin-8; IL-13: interleukin-13; IL-35: interleukin-35; IL-33: interleukin-33; IL-11: interleukin-11.
The immune microenvironment is a hallmark of cancer, and the tumor immune microenvironment is more complex than the psoriatic immune microenvironment [14]. The tumor immune microenvironment includes a set of cell types, such as endothelial cells, pericytes, immune inflammatory cells, and cancer-associated fibroblasts [17]. These cells secrete IL-6, IL-23, IL-22, IL-17, IL-1β, and TGF-β, causing cancer cell proliferation. Moreover, as the foundation of cancer, proliferative cancer cells also produce IL-6, IL-11, and other cytokines, leading to malignant proliferation and producing matrix metalloproteinases (MMPs), VEGF, and other adhesion factors, which promote angiogenesis for the nutrition supplementation of tumor cell proliferation and tumor cell circulation, as well as immune cell recruitment and activation [18][19][18,19]. Ultimately, the tumor immune microenvironment and proliferative cancer cells form a positive loop, driving tumor progression forward.
2.2. Metabolic Reprogramming
Metabolic reprogramming is another hallmark of cancer. The metabolic pattern of starved rapidly proliferating tumor cells changes in order to meet nutritional requirements [20]. Tumor cell proliferation mainly depends on glycolysis, referred to as the Warburg effect. To maintain the Warburg effect, the expression of glucose transporter 1 (GLUT1), pyruvate kinase (PK), hexokinase (HK), and phosphoglycerate mutase (PGAM) increases in tumors. Meanwhile, the TCA cycle is reprogrammed by the upregulation and mutation of fumarate hydratase (FH), succinate dehydrogenases (SDHs), and isocitrate dehydrogenase 1/2 (IDH1/2). In addition, the changes in enzymes such as glutamine transporter, glutaminase (GLS1), arginine succinate synthase (ASS1), phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), serine hydroxymethyltransferase 1/2 (SHMT1/2), and indoleamine 2, 3-dioxygenase 1/2 (IDO1/2) allows for the metabolic reprogramming of amino acids such as glutamine, arginine, serine, and tryptophan metabolic pathways. Sphingomyelin metabolites are also involved in tumor development [21]. In summary, the glucose, amino acid, and lipid metabolisms are reprogrammed in cancer.
The glucose, amino acid, and lipid metabolisms also undergo metabolic reprogramming in psoriasis patients. Hyperproliferative keratinocytes are subjected to the Warburg effect. GLUT1 is required for glucose uptake by proliferative keratinocytes, and its expression is increased in hyperproliferative keratinocytes [22]. Pyruvate kinase isozyme type M2 (PKM2) is generally increased in psoriasis patients and proliferates keratinocytes to enhance glycolysis [23]. IL-17 downregulates protein phosphatase 6 (PP6) and induces the generation of arginase-1 (ARG1) in psoriasis [24]. The deficiency of PP6 leads to the accumulation of AGR1, promoting skin inflammation by driving polyamine production. The IL-17A/MALT1/c-Jun axis induces the abnormal expression of GLS1 in immune cells and keratinocytes [25]. Sphingolipid metabolite sphingosine-1-phosphate (S1P) is increased in psoriasis patients [26].
2.3. Epigenetic Reprogramming
Epigenetics refers to heritable molecular processes whereby a constant DNA sequence produces variable gene expression patterns without any mutational change in the DNA sequence. Compared to normal cells, tumor cells exhibit epigenetic changes such as DNA methylation, histone modifications, and genome-wide changes in the three-dimensional (3D) chromatin structure representing epigenetic mutations [27][28][27,28]. In tumor cells, alterations in the activity of epigenetic-related proteins such as histone deacetylases (HDACs), bromodomain proteins (BRDs), TET protein demethylases, histone lysine methyltransferases (HMTs), and lysine-specific demethylases (LSDs) result in abnormal histone acetylation and methylation; DNA methyltransferases (DNMT) aberrantly affect DNA/RNA methylation [27][28][27,28]. These epigenetic reprogrammings cause features such as excessive cell proliferation, death resistance, immune escape, genomic instability, and microbiome polymorphisms.
In psoriasis, epigenetic reprogramming occurs by affecting enzymes such as HADCs, BRDs, and HMTs, resulting in abnormal histone acetylation and methylation, as well as abnormal DNA methylation. It has been reported that global DNA methylation is increased in psoriasis patients, and genes with hypermethylated promoters enrich gene ontology processes, such as cell development and differentiation, actin cytoskeleton organization, cell adhesion, and motility; meanwhile, genes with hypomethylated promoters enrich immune activation processes, including inflammation, T cell activation, cytokine production, and cell proliferation [29][30][29,30]. Methylation modifications at CpG sites are increased in psoriatic lesions [31]. The promoter of the p16INK4a gene is methylated in the epidermis of psoriasis patients, and p16INK4a mRNA expression is also elevated, with high degrees of methylation of the promoter region and hypomethylation of some other regions also occurring in CD4+ cells [32]. Enhancer homolog 2 of the Zeste gene (EZH2) is a histone H3K27 methylase, whereas EZH2 acts on kallikrein-8 (KLK8) to cause the abnormal proliferation of keratinocytes in psoriasis [32].