Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Evgeny E. Bezsonov and Version 2 by Conner Chen.
Cardiovascular disease (CVD) is the main cause of global death, highlighting the fact that conventional therapeutic approaches for the treatment of CVD patients are insufficient, and there is a need to develop new therapeutic approaches. In recent years, decoy technology, decoy oligodeoxynucleotides (ODN), and decoy peptides show promising results for the future treatment of CVDs. Decoy ODN inhibits transcription by binding to the transcriptional factor, while decoy peptide neutralizes receptors by binding to the ligands.
- cardiovascular diseases
- decoy
- oligodeoxynucleotide
- peptide
12. Decoys
A decoy is defined as something that is aimed to draw attention away from a particular situation or from an intended course of action. This is the premise of decoy base therapy where the drug candidates represent an opportunity to inhibit a known activated regulatory pathway that promotes CVD. Decoy technology includes two main approaches, decoy ODN and decoy peptides [1][2][9,10].
Decoy ODN is a DNA construct that regulates the expression of genes related to disease through a reduction of authentic cis–trans interaction. Transcription factors, as trans-regulatory elements, regulate gene transcription by binding to the cis-element [3][11]. Decoy ODNs have a similar structure to the cis-element of the target gene and trap the transcription factor and modulate the gene expression (Figure 1A) [1][9].
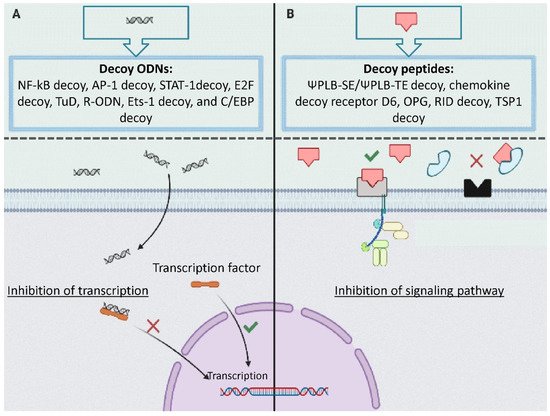
Figure 1. The mechanism of action of decoys: (A) decoy oligodeoxynucleotides (ODN) including NF-kB decoy, AP-1 decoy, STAT-1decoy, E2F decoy, tough decoy, R-ODN, and C/EBP decoy inhibits transcription by binding to the transcriptional factor; (B) decoy peptide including ΨPLB-SE/ΨPLB-TE decoy, chemokine decoy receptor D, decoy receptor osteoprotegerin neutralizes receptors by binding to the ligands.
Many pathological processes related to CVD such as inflammation are triggered by associated ligand–receptor interactions [4][12]. Peptide decoys, also known as decoy receptors, are proteins that have a similar structure to receptors. Peptide decoys can bind to specific ligands and trap them resulting in attenuated ligand–receptor interactions (Figure 1B).
Delivery of Decoy
The efficacy of decoys mostly relies on their stability and delivery. Local delivery is considered the most effective approach for overcoming systemic administration problems; however, local delivery is only possible for a few organs such as the lungs and the eyes [5][13]. Generally, in systemic delivery, most mammalian cells could take up sufficient decoys; however, to maximize cellular uptake and increasing efficiency under physiological conditions, active transport delivery systems and/or receptor-mediated endocytosis should be considered [5][13]. Moreover, decoy delivery can also be regarded as passive (for tumor and other highly permeable organs) or active targeting delivery (for peripheral tissues) [6][14].
Various delivery systems for decoys may be employed, including electrically enhanced transfer, pressure-mediated transfer, biolistic bombardment, lipidoids, cationic liposomes, lipid-based nanoparticles, virus of Japan (HVJ) liposomes (the delivery time is approximately 15–30 min and sustainability up to 1–2 weeks), bacterial vectors, hemagglutinating microsphere-aided delivery, steroid mediated gene transfer, peptide-mediated delivery, and aptamer/oligonucleotide chimeras, exosomes and cell-based carriers [5][13]. For in vivo delivery, liposomes and viral vectors such as adenoviruses, adeno-associated virus (AAV), and lentivirus are mostly used in mammalian cells [6][14]. The non-viral delivery systems can be further divided into polymer, lipid, protein, and peptide-based delivery methods [5][13]. Poly(D, L-lactide co-glycolide) and chitosan), gelatin, liposomes, and lipid-based nanoparticles, palmitoyl-oleyl-phosphatidylcholine (POPC), poly(glycoamidoamine)s, and biodegradable polymer d,l lactide-co-glycolide (PLGA) are some examples of carriers for decoy delivery [5][13].
2. Effects of Decoys on Non-Atherosclerotic CVD
2.1. Nuclear Factor-Kappa B
The nuclear factor-B (NF-κB) family mediates the processes of inflammation, cell differentiation, and proliferation, as well as cellular response regulation of hypoxia, stretch, stress, and ischemia. Intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule (VCAM-1), and endothelial leukocyte adhesion molecule-1 (ELAM-1) adhesion molecules are upregulated by the transcription factor NF-κB. NF-κB is implicated in several cardiovascular diseases including myocardial ischemia, myocardial reperfusion injury, atherosclerosis, ischemic preconditioning, cardiac hypertrophy vein graft disease, and cardiac failure [7][61].
Thrombosis results in vessel occlusion and contributes to the pathogenesis of cardiovascular diseases. The tissue factor is a glycoprotein that has an integral role in the triggering of blood coagulation through the extrinsic pathway and acts as a receptor for coagulation factor VII and activated factor VII that could initiate the coagulation cascades; therefore, tissue factor can promote thrombosis in coronary heart disease. Wang et al. used a decoy to bind or “trap” NF-κB and prevent the transcription of the tissue factor gene. In one study, human umbilical cord vein endothelial cells (HUVECs) were transfected with NF-κB transcription factor decoy oligodeoxynucleotides (TFD) using liposomes, and the results showed that the NF-κB decoy competed with the endogenous NF-κB site sequence in the tissue factor promoter for binding to transcription factor NF-κB to block expression of the tissue factor gene [8][20].
NF-κB regulation may suppress abdominal aortic aneurysm (AAA) progression, and inhibition of NF-κB can protect against AAA development. Akimoto et al. developed an NF- kB decoy to inhibit NF-κB and delivered this decoy to AAA-induced rats via a bioabsorbable sheet. The bioabsorbable sheet delivered the decoy into the target tissues, and treatment resulted in the decoy decreasing the size of the aneurysm, compared with the controls [9][21].
NF-κB modulates gene transcription involved in myocardial ischemia-reperfusion injury. After extended myocardial preservation blocking NF-κB reduces ischemia-reperfusion injury and improves cardiac function. Sakaguchi et al. transferred NF-κB decoy into rat hearts via a hemagglutinating virus of Japan (HVJ)-liposome vector. NF-κB decoy therapy enhanced the recovery of left ventricular function by lowering serum creatine phosphokinase, neutrophil infiltration, tissue IL-8, and myocardial water content, compared to the control group [10][22].
NF-κB activates cytokines, adhesion molecule genes, and inducible NO synthase (iNOS). Inflammation underlies myocarditis pathogenesis; therefore, Yokoseki et al. developed a decoy against the cis-element of NF-κB to inhibit the progression of experimental autoimmune myocarditis (EAM) and infused decoy into the rat coronary artery. Their results showed that treatment lowered ratios of myocarditis-affected areas to the ventricular cross-sectional area and decreased expression of ICAM-1, iNOS, IL-2, and TNF in the myocardium [11][23].
Acute transplant rejection or graft arteriopathy may complicate cardiac transplantation and attenuate survival. NF-κB is responsible for gene transcription of inflammatory or proliferative mediators. Suzuki et al. used an HVJ-AVE-liposome to inject an intraluminal NF-κB decoy into cardiac allografts of a murine model. They showed acute rejection of allografts from the major mismatch group, while the NF-κB decoy enhanced graft survival. Migration of cells, the thickness of intima, and inflammatory factors were increased in control allografts of the minor mismatch group, which were decreased by the NF-κB decoy. Treatment by decoy reduced cell infiltration, as well as ICAM-1, VCAM-1, NF-κB, and major histocompatibility complex (MHC) class I and II in allograft myocardium [12][24].
Sawa et al. used an NF-κB decoy for the attenuation of ischemia-reperfusion injury in the myocardium. NF-κB decoy was transferred to the rat hearts by coronary infusion of HVJ-liposome at the time of the cardioplegic arrest. The percentages of recovery of left ventricular pressure and coronary flow were higher in the NF-κB decoy group when exposed to ischemia and reperfusion. Neutrophil adherence to endothelial cells and levels of interleukin-8 were lower in the treatment group [13][25].
Morishita evaluated the effect of an NF-κB decoy as an effective therapy for myocardial infarction in rats. The results showed that in vivo transfer of NF-κB decoy before and after infarction, before occlusion of the coronary artery, or immediately after reperfusion decreased the myocardial infarction area. Furthermore, decoy therapy reduced the expression of cytokines (IL-6 and IL-8) and adhesion molecules (VCAM and ICAM) [14][26].
Nakashima et al. designed a novel chimeric decoy strategy to inhibit NF-B and Ets simultaneously for the treatment of AAA. Chimeric decoy inhibited AAA progression, decreased aneurysmal dilation, reduced MMP expression, and inhibited macrophage migration. Furthermore, it inhibited elastin fiber destruction in the aorta. Importantly, the chimeric decoy ODN inhibited aneurysmal dilatation more than the NF-B decoy [15][27].
Kalinowski et al. transfected an NF-κB decoy into hypercholesterolemic rabbits in order to prevent restenosis following balloon angioplasty. The NF-κB decoy inhibited proliferation of VSMC in vitro; however, the reduction of the neointimal area by the NF-κB decoy did not differ, compared to the control group [16][28].
By use of genetically engineered adenoviral (Ad) vectors, bone marrow-derived dendritic cells (DCs) expressed immunosuppressive molecules for promoting T cell unresponsiveness. NF-κB decoy treatment can suppress the maturation of DC through the NF-κB pathway. Bonham et al. used the combination of an NF-κB decoy and rAd vectors encoding CTLA4-Ig (Ad CTLA4-Ig) to produce stably immature murine myeloid DCs that secreted the co-stimulation blocking agent. The capacity of these DCs for stimulation was disrupted resulting in enhanced apoptosis of T cells. Moreover, administration of these DCs before transplantation prolonged survival of MHC-mismatched vascularized heart allografts [17][29].
Zhang et al. examined whether an NF-κB decoy could further augment the inhibition of cellular proliferation by radiation of VSMCs in vitro. The irradiation promotes activation or nuclear translocation of NF-κB in VSMCs and transfection of the NF-κB decoy inhibited the radiation-induced NF-κB activation in VSMCs. Transcription and translocation of NF-κB downstream molecules including ICAM, iNOS, and TNF-α were reduced. Inhibition of NF-κB by decoy increased apoptosis and reduced proliferation and survival in the irradiated VSMCs [18][30].
Miyake et al. investigated the effect of a chimeric NF-κB and Ets decoy on aortic dilatation in a rabbit AAA model. Results showed that the chimeric decoy inhibited the aortic dilatation progression that was confirmed histologically. The chimeric decoy also decreased the activities of MMP-2 and MMP-9 and inhibited elastin proteolysis. Furthermore, suppressing VCAM-1 and MCP-1 gene expression by the chimeric decoy inhibited macrophage infiltration in the media and adventitia [19][31].
DC can modulate immune responses by altering the transcription of NF-κB-regulated genes of surface costimulatory molecules (CD80, CD40, CD86). Tolerance to DC has been related to impaired NF-κB-dependent transcription of costimulatory genes and NF-κB translocation to the nucleus [20][32]. Giannoukakis et al. demonstrated that an NF-κB decoy was incorporated by bone-marrow-derived DC and the decoy inhibited LPS-induced nitric oxide production, which is a maturation marker of DCs. Treatment by this decoy repressed the cell-surface expression of CDs without affecting MHCs expression. Additionally, decoy treatment inhibited the production of the Th1- type cytokine. Importantly, infusion of the NF-κB decoy DC into allogeneic recipients before heart transplantation prolonged allograft survival in the absence of immunosuppression [20][32].
Myocardial reperfusion injury is the result of inflammation following ischemia that allows subacute polymorphonuclear leukocytes (PMNs) adhesion, for example, via NF-κB activation. Kupatt et al. studied the effect of targeted NF-κB decoy treatment in the area at risk (AAR) on the size of infarct and regional myocardial function. Liposomes containing NF-κB ODN were retroinfused into the anterior interventricular vein in pigs. NF-κB decoy retroinfusion decreased infarct size, while functional reserve of the AAR tended to improve; therefore, the NF-κB decoy provided postischemic cardioprotection in pig hearts [21][33].
NF-κB mediates the vascular response to injury, though its role in the mechanism of in-stent restenosis has not been clarified [22][34]. Ohtani et al. tested the blockade of NF-κB by stent-based delivery of an NF-κB decoy for the reduction of in-stent neointimal formation in rabbits with hypercholesterolemia. Results showed that infiltration of monocytes and expression of MCP-1 were reduced by the NF-κB decoy, while CD14 activation on circulating leukocytes was suppressed by the decoy. Importantly, the neointimal formation was attenuated by the NF-κB decoy. Moreover, transfection of the NF-κB decoy inhibited the proliferation of human coronary artery SMCs in vitro [22][34].
Miyake et al. developed a modified chimeric decoy against NF-κB and Ets as a novel therapeutic approach for AAA. The decoy had a ribbon-shaped circular structure that had not been chemically modified to maximize its resistance against endonuclease for systemic administration. Ribbon-type decoy ODN (R-ODNs) was administrated intraperitoneally in an elastase-induced rat AAA model. Interestingly, R-ODN inhibited aortic dilatation, although a conventional phosphorothioate chimeric decoy ODN (PS-ODN) failed to prevent the aneurysmal development. Moreover, R-ODN inhibited the activation of MMP-12 and MMP-9 in the aneurysm wall and suppressed the secretion of cathepsin K and B from macrophages, but it did not inhibit the recruitment of macrophages. Chimeric R-ODN also prevented aortic dilatation, while aneurysm development was not prevented by conventional phosphorothioate decoy ODN treatment [23][39].
Kimura et al. studied the activity of NF-κB in a rat model of PAH. They showed that the NP-mediated NF-κB decoy delivery into lungs prevented monocrotaline-induced NF-κB activation. Blockade of NF-κB by NP-mediated delivery of the NF-κB decoy attenuated inflammation and proliferation, attenuating the development of PAH and pulmonary arterial remodeling induced by monocrotaline [24][59].
Aoik et al. examine the regressive effect of a chimeric decoy, which simultaneously inhibited NF-κB and Ets-1, on IA development in the rat model [25][38]. They had previously investigated the expression and role of Ets-1 in CA development using decoy ODN. Treatment with Ets decoy oligodeoxynucleotides resulted in the prevention of CA enlargement, upregulation of MCP-1 expression, and increase in macrophage accumulation in CA walls [26][41]. Chimeric decoy ODNs decreased intracranial aneurysm (IA) size and thickened IA walls of preexisting IAs induced in the rat model, although the treatment with NF-κB decoy ODNs failed to regress preexisting IAs. Chimeric decoy ODN-treated rats exhibited decreased expression of monocyte chemotactic protein-1 and macrophage infiltration in IA walls [25][38]. Aoik et al. also showed that treatment with NF-κB decoy restored the reduced expression of procollagen type I, III, and LOX [27][62]. This group showed that NF-κB decoy prevented CA formation when it was administered at the early stage of aneurysm formation in rats. Macrophage infiltration and expression of downstream genes were dramatically inhibited by NF-κB decoy oligodeoxynucleotide [28][35].
Miyake et al. used chimeric decoy containing consensus sequences of both NF-κB and Ets binding sites to treat AAA. Inhibitory effects of chimeric decoy on MMP-1 and -9 expressions were confirmed by ex vivo experiments using a human aorta organ culture. Importantly, treatment with chimeric decoy ODNs significantly decreased the size of AAA [29][37].
Shiraya et al. showed that transfection of a chimeric decoy inhibited aortic dilatation, both in normotensive and hypertensive rats. Destruction of elastic fibers was also inhibited by transfection of the chimeric decoy in both hypertensive rats and normotensive rats. The expression of MMP-2, -3, -9, and -12, as well as the intercellular adhesion molecule, was significantly attenuated by the chimeric decoy ODN, accompanied by inhibition of the migration of macrophages [30][36].
2.2. Activator Protein-1 (AP-1)
AP-1 is a transcription factor involved in the transcriptional regulation of genes implicated in cell proliferation and extracellular matrix production in response to inflammatory cytokines and oxidative stress [31][42].
Transplant vasculopathy (TV) resulting from cardiac transplantation results in obstructive lesions in vessels. Activation of AP-1 increases the migration and proliferation of SMC. Remes et al. examined AAV-mediated delivery of an RNA hairpin AP-1 decoy oligonucleotide for the treatment of TV in a mouse aortic allograft model of TV. AP-1 decoy oligonucleotides were expressed in the cells of graft tissue. Ex-plantation after 30 days and evaluation showed that AP-1 decoy oligonucleotide therapy reduced the intima-to-media ratio in the grafts by 41.5 percent. Additionally, in treated grafts, expression of cytokines and adhesion molecules were greatly decreased, as were MMP-9-positive cells, SMCs, and inflammatory cell infiltration [31][42].
Infusion of decoy oligodeoxynucleotides against the activator protein-1 (AP-1) binding site (dec-ODN) prevents neointimal proliferation and thickening [32][43]. Lin et al. delivered a decoy against AP-1 in-stent and evaluated the inhibitory effects on restenosis. Decoy drug-eluting stents (DESs) were implanted and were compared to normally implanted stents in the abdominal aorta of rabbits. The decoy transmitted in-stent inhibited TGF-1 and CTGF expression, as well as preventing neointimal thickening and restenosis eight weeks after stent implantation. Re-endothelialization was not affected by the decoy [32][43].
Endothelin-1 synthesis in vessel walls is a powerful co-mitogen for VSMCs stimulating restenosis following percutaneous transluminal coronary angioplasty (PTCA). Deformation-induced expression of prepro-endothelin-1 is governed by AP-1. Buchwald et al. developed a decoy against AP-1 that was given into the coronary arteries of hypercholesterolemic minipigs during PTCA. Decoy treatment decreased neointimal development in coronary arteries, which was correlated with a reduction in AP-1 nuclear translocation and endothelin-1synthesis in the vessel wall [33][44].
Arif et al. reduced aortic elastolysis through decreased MMP expression with decoy ODNs neutralizing AP-1. Exposure to AP-1 neutralizing dODNs resulted in a significant reduction of basal and interleukin-1β-stimulated MMP expression and activity in mAoSMCs. Moreover, increased migration and formation of superoxide radical anions were substantially decreased in mAoSMCs by AP-1 dODN treatment [34][45].
2.3. Signal Transducer and Activator of Transcription-1 (STAT-1)
STATs proteins are downstream of the Janus kinases (JAKs) signaling pathway. Dysregulation of JAK-STAT signaling has been shown to be related to cardiovascular diseases [35][63].
Acute myocardial rejection is notable by enhanced leukocyte migration into the graft myocardial tissue. AP-1 and STAT-1 are transcription factors that regulate the expression of vascular adhesion molecules and therefore very important in this process. Hölschermann et al. investigated the effect of a decoy ODN that targeted transcription factors AP-1 and STAT-1 on acute cardiac allograft rejection in a rat transplantation model. They transplanted Wistar–Furth cardiac allografts into Lewis rats following perfusion with AP-1 or STAT-1 dODN solution (5 μmol/L). Treatment with AP-1 and STAT-1 decoys extended cardiac allograft survival by approximately 40%. Immunohistochemical results showed a significant reduction of infiltrating leukocytes, specifically T cells. Furthermore, the expression of ICAM-1 and VCAM-1 in the endothelium was found to be noticeably reduced by decoys. Therefore, both STAT-1 and AP-1 decoys suppressed graft endothelial adhesion molecule expression, diminished graft infiltration, and delayed acute rejection substantially [36][46].
Stojanovic et al. assessed the STAT-1 decoy in heterotopic mouse heart transplantation without immunosuppression. Mouse heart allografts vessels were pre-treated with STAT-1 decoy. A single treatment with STAT-1 decoy decreased both rejection scores by 85% and 70% in dODN-treated allografts. Furthermore, graft myocardium infiltration by monocyte and T cells was reduced. In addition, STAT-1 decoy impaired pro-inflammatory gene expression greater than 80% 24 h post-transplantation, but the signal was lost 9 days after transplantation [37][47].