Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Daria Bortolotti and Version 2 by Catherine Yang.
Human herpesviruses (HHVs) are highly widespread among humans and therefore are among the pathogens most responsible for gestational infections. HHVs are classified into three subfamilies (alpha-, beta- and gammaherpesvirinae), and they are able to establish permanent latency within the host in specific cells. The alphaherpesvirinae family includes herpes simplex type-1 (HSV-1 or HHV-1), herpes simplex type-2 (HSV-2 or HHV-2) and varicella zoster virus (VZV or HHV-3). The betaherpesvirinae family includes cytomegalovirus (CMV or HHV-5), HHV-6A/B and HHV-7. The gammaherpesvirinae family consists of Epstein–Barr virus (EBV or HHV-4) and Kaposi’s sarcoma-associated herpesvirus (KSHV or HHV-8).
- viruses
- immune system
- DNA viruses
- RNA viruses
1. HSV-1 and HSV-2
Normally, HSV-1 predominates in orofacial lesions and typically is found in the trigeminal ganglia, while HSV-2 is most found in the lumbosacral ganglia. The greatest risk of disease in the newborn is represented by the late-pregnancy infection of genitals in a previously unexposed woman, while recurrent infections are rarely associated with disseminated neonatal disease in immune-competent women.
However, a primary HSV infection of a pregnant woman leads to greater risks for both mother and child. Although HSV-infected pregnant women have rare or no clinical recurrences, there is still the risk of intrapartum transmission [1][114].
Women who already has antibodies to both HSV-1 and HSV-2 at the onset of pregnancy, which is the most common condition, have the least risk of perinatal transmission [2][115]. On the contrary, new-onset HSVs infection occurring late in pregnancy carries a 30% to 50% risk of neonatal infection, while early pregnancy infection carries a risk of less than 1% [3][116]. The possible explanation could be that when primary HSVs infection occurs during late pregnancy, the time for developing specific antibodies and suppressing viral replication before labor is not enough.
Again, while HSV-1 transmission from mother to newborn seems to be easier in the presence of both primary infection or recurrences [4][117], primary HSV-2 infection transmission to the fetus is less frequent [5][118], but it has been associated with a higher incidence of preterm birth [6][119] (Figure 12). The clinical manifestations of neonatal HSVs infection include encephalitis and disseminated disease, with a mortality rate of more than 50%. Survivors are, however, compromised, usually with significant neurologic deficits, blindness, seizures and learning disabilities (Figure 12). Different studies have also stated that HSVs infection can affect maternal immune responses, resulting in loss of HLA-G [7][120], cell death and reduced human chorionic gonadotropin (HCG) secretion [8][121] (Figure 12). These changes in trophoblast function could explain why both HSV-1 and HSV-2 have been associated with spontaneous pregnancy loss [9][122] and IUGR pregnancies [10][11][85,123] (Figure 12).
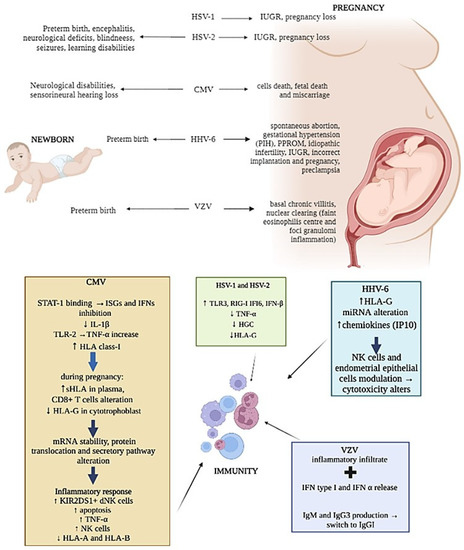
Figure 12.
Representation of herpesviruses infection’s main effects on newborn, pregnancy and mother immunity.
Moreover, HSV is capable of increasing the expression of TLR3, RIG-I, IFI6 and IFN-β proteins as well as decreasing TNFα production in terms of human placental explant cultures [12][124] (Figure 12), affecting maternal innate immune system antiviral responses.
2. CMV
Among herpesviruses, CMV is one of the most vertically transmitted and it represents the most common cause of congenital infection in high-income countries, causing neurological disability and sensorineural hearing loss in newborns [13][125] (Figure 12). The intrauterine infection caused by CMV occurs in 0.3% to 2.3% of births [14][126]. CMV intrauterine transmission is more common after primary infection (30–40%) than after non-primary infection (1%) [15][16][127,128]. Nevertheless, it was estimated that non-primary maternal infections are responsible for the majority of congenital CMV infections [17][129].
CMV, such as many other viruses, employs multiple mechanisms to exploit the vulnerability of the placenta and impair the innate host response in order to spread the infection, including the expression of several viral proteins such as IE1 [18][130], IE86 [19][131], UL44, IE2 and UL94 [18][20][21][130,132,133].
The effect of CMV on the placenta has been confirmed by proving that ultraviolet-inactivated human CMV leads to syncytiotrophoblast apoptosis via TLR2, also increasing TNFα production [22][23][134,135]. Interestingly, in human villous explants, CMV did not induce the expression of RIG-I and MDA5 proteins and cytokine production [24][136], suggesting that RLRs could play a central role in the inhibition of vertical transmission of the virus as well as in the selectivity of vertical transmission of viruses across the placenta.
CMV can modulate the expression of HLA molecules by encoding specific viral proteins, mainly decreasing their expression on the cell surface to prevent immune cells recognition. In addition, CMV can selectively up-regulate specific HLA class-I molecules [25][137] (Figure 12). As aforementioned, HLA-G is physiologically expressed at the maternal–fetal interface on trophoblasts and is one of the major molecules targeted by viruses during pregnancy [26][138]. The concentrations of sHLA-G normally increases in the plasma of pregnant women during the first trimester of pregnancy [27][139], but during CMV infection, a reduction in HLA-G in cytotrophoblasts was observed [28][140] together with its up-regulation in peripheral blood cells [29][141] (Figure 12). This effect is due to the interaction of specific CMV proteins with the HLA-G promoter, which affects mRNA stability and protein translation and secretion [30][31][32][142,143,144] (Figure 12).
Several studies have reported the failure of dNK to control CMV infection. Since NK cells activity is regulated by the expression of activation/inhibitory killer imuunoglubulin-like receptors (KIRs), such as KIR2DL4, KIR2DS1/5, etc. as activating, and KIR2DL1/2/3, etc. as inhibiting, the expression of these receptors and of their ligands on target cells can be exploited by viruses as immune-escape mechanisms. Crespo et al. [33][145] demonstrate that CMV infection of human HLA-C2 + decidual stromal cells drives the cytotoxic activation of dNK and placental NK (pNK) cells in vitro by engaging KIR2DS2, and that KIR2DS1 or KIR2DS5-negative pregnant women have a lower ability to control placental CMV infection, developing complications. Van der Ploeg et al. [34][21] reported the molecular basis for the increased degranulation response of KIR2DS1 + dNK to CMV infection (Figure 12). Yan et al. [35][146] show that a KIR2DL4/HLA-G combination induces high NK cytotoxicity, which might be beneficial uterine CMV infection. Other studies described the presence of adaptive NK cell expansion found during different viral infections, concluding that in CMV-infected individuals, adaptive NK cells may be established probably as the result of opportunistic viral reactivation [36][147].
Moreover, the interaction between CMV and dNK cells can be the cause fetal death or miscarriage due to NK cell cytotoxic activity [37][148] (Figure 12). However, the CMV infection of EVT did not diminish the ability of EVT to increase FOXP3+ and PD1HI T-regs [38][149], suggesting that its infection does not alter the capacity of EVT to promote immune tolerance. This finding confirms the observation that dNK fails to degranulate in response to CMV-infected EVT, thus also maintaining immune tolerance in the presence of infection [39][20].
Moreover, the failure of dNK to respond to CMV-infected EVT during in vitro co-culture [39][20] may leave decidual CD8+ T cells as the predominant effector cell to clear pathogen-infected EVT.
Seropositive women during late pregnancy demonstrated an accumulation of highly differentiated CMV-specific T cells [40][150]. In fact, CMV seropositivity was shown to dramatically alter the maternal CD8+ T-cell repertoire during pregnancy [40][150], and T-cell responses to CMV rely heavily on HLA-C-restricted signals [41][151] (Figure 12). CMV CD8+ T cells were also found increased particularly in decidual tissue and were found able to produce IFNγ and restricted to recognizing viral peptides presented by HLA-A or HLA-B molecules, limiting the spread of infection to trophoblasts and/or the fetus [42][152].
3. HHV-6
HHV-6 is widely spread during pregnancy as well. HHV-6 DNA has been detected in blood and tissue samples from women with several types of gestational problems, including spontaneous abortions, gestational hypertension and preterm birth, in association with the detection of high anti-HHV-6 IgM and IgG titers [43][153] (Figure 12). HHV-6 DNA was also found in the amniotic fluid of women with gestational complications [44][154], as pregnancy induced hypertension (PIH) and the premature preterm rupture of membranes (PPROM) (Figure 12).
To date, despite different congenital herpetic infections having been associated with late IUGR, no direct implication of HHV-6 infection has been reported. In particular, HLA-G expression and HHV-6 infection have been evaluated in placentas from late-onset IUGR newborns compared to placentas from uncomplicated pregnancies [45][155], since HHV-6 is known to exploit the modulation of HLA-G as an immune-escape mechanism.
HLA-G increased and HHV-6 presence were found to correlate in IUGR placenta samples [45][155]. These preliminary results underline a direct relationship between HHV-6 infection and HLA-G deregulation that might affect vessel remodeling and prevent the correct pregnancy outcome in the IUGR condition (Figure 12).
However, HHV6 is comprised of two species, HHV-6A and HHV-6B [46][47][156,157]. While most of the population is infected by HHV-6B by 2 years of age, HHV-6A infection usually occurs later [48][49][50][158,159,160]. In particular, HHV-6A clinical manifestations are still unclear, but the presence of HHV-6A in endometrial epithelial cells of a subgroup of idiopathic infertile women [51][161] supported the role of HHV-6A [52][162] (Figure 12).
Moreover, it has been observed that HHV-6A infection induces a profound remodulation of miRNA expression in human cells of different origin [53][163], including human endometrial cells, in which HHV-6A modulates at least 16 miRNAs with potentially critical roles during embryo implantation [54][164]. These virus-induced alterations in the miRNA expression of endometrial cells might affect trophoblast cell behavior (Figure 12), supporting the hypothesis that HHV-6A might be associated with interference in correct implantation and pregnancy outcome [55][165].
The abilities of NK and endometrial cells have been described to be changed by HHV-6A infection. In fact, phenotypical and functional modifications of both endometrial NK (eNK) and epithelial cells have been reported in HHV-6A-positive infertile women samples, suggesting an imprint due to HHV-6A infection on both eNK cell immune-phenotype and receptors repertoire (Figure 12). In particular, during HHV-6A infection, eNK cells seem to acquire a cytotoxic profile as an attempt to limit the infection, which involves the NKG2D receptor [52][162]. The persistence of activated eNK and of subclinical HHV-6A infection could alter endometrial environment and disadvantage embryo implantation and placentation, and it could potentially have serious adverse side effects, such as pre-eclampsia, fetal growth restriction and stillbirth, as demonstrated by the increase in chemokines, mainly IP10 and FasL, in uterine flushing samples from HHV-6A-positive infertile women (Figure 12).
In addition, Rizzo et al. observed a lower percentage of KIR2DL4-positive eNK cells in primary infertile women in correlation to the diminished expression of soluble HLA-G [56][166]. This evidence supports the potential role of HHV-6 in female diseases, as a consequence of HLA-G modulation, that can in turn induce anergy to eNK cells via the inhibitory KIR2DL4 receptor [56][166].
4. VZV
VZV is the etiological agent for chicken pox at time of primary infection, and it is usually associated to mild clinical course, but in pregnant women, it may occasionally lead to serious maternal and fetal diseases. Maternal VZV can infect the baby by different routes: (a) transplacental viremia, (b) ascending infection during birth or (c) respiratory droplet/direct contact with infectious lesions after birth.
Even if the primary mechanism of VZV transfer across the placenta remains unclear, it is postulated that infected T cells might be present in the decidua basalis [57][4], where both CD4+ and CD8+ T cells are reprogrammed by the virus, becoming more capable of crossing into the intervillous space [58][167].
However, reports vary on the histological features of VZV placental infection, suggesting that VZV could be transmitted to the fetus via the placenta without apparent viral replication within the placenta [59][168] (Figure 12).
Interestingly, nearly 20% of infants with intrauterine-acquired VZV primary infection develop neonatal or infantile zoster, usually with uncomplicated course [60][169]. The disease is thought to represent reactivation of the virus after primary infection in utero, and the short viral latency may be explained by the immature cell-mediated immune response in young children.
Moreover, recurrent chickenpox has been documented in pregnant women [61][170], underlining again the key role of the immune system.
In addition to the tests of general antibody reactivity, tests of antibody avidity [62][171] and IgG isotype [63][172] can be used to assess the nature of VZV antibody responses. The avidity of antibodies seems to increase thereafter and during shingles, while there is a switch from IgM and IgG3 to IgG1 after primary disease [64][173] (Figure 12). Therefore, the clinical manifestation of these pregnant women can be due to the high virus load and low immune responses [65][174], whether the effect of pregnancy and associated hormones on VZV replication is not known.
VZV infection causes a very early release of IFN type I, which is particularly abundant at the lesion level [66][175] (Figure 12). NK cells can also be found early after VZV infection [67][176], suggesting their central role in controlling viral spread. In fact, while both cytotoxic NK and primed CD8+ T cells were nearly absent during the early phase of life-threatening primary VZV infection [68][177], their responses to VZV seem to be protective and associated to mild symptoms [69][178]. As an example, the detection of T cells within three days after the appearance of the varicella rash, with rapid host response to primary VZV infection, has been known to be associated with milder rash and a more rapid clearance of viremia in healthy subjects [67][176].