Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Anton R Kiselev.
Non-alcoholic fatty liver disease (NAFLD) and arterial hypertension (AH) are widespread noncommunicable diseases in the global population. Since hypertension and NAFLD are diseases associated with metabolic syndrome, they are often comorbid. In fact, many contemporary published studies confirm the association of these diseases with each other, regardless of whether other metabolic factors, such as obesity, dyslipidemia, and type 2 diabetes mellites, are present.
- non-alcoholic fatty liver disease
- hypertension
- steatohepatitis
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) and arterial hypertension (AH) are common noncommunicable diseases in the global population. To date, the impact of NAFLD extends far beyond the liver; its association with an increased risk of cardiovascular disease (CVD), both in isolation and as part of the metabolic syndrome, has been proven [1]. The relationship between CVD and NAFLD is also explained by the commonality of risk factors underlying the development of these diseases. In addition to already known risk factors (abdominal obesity [2], high blood cholesterol [3], metabolic disorders [4], etc.), data have been obtained on other factors that can trigger the development and progression of both NAFLD and CVD [5]. Among these, we should mention an increase in serum levels of uric acid [6], C-reactive protein, interleukin (IL)-6 [7], fibrinogen, von Willebrand factor, and plasminogen activator inhibitor-1 [8], as well as an augmented thickness of epicardial fat [9] and intima-media complex [10]. AH is included in the criteria for metabolic syndrome [11]. It accompanies NAFLD quite frequently and is a major risk factor for CVD [12].
Despite the presence of bidirectional relationship between NAFLD and AH, it is often difficult to establish which of the two conditions occurs first. However, the existence of common mechanisms determines the high risk of developing one of them in cases where the other is already present. The mechanisms of the relationship between NAFLD and AH are mostly known (Figure 1) [70][13], but they continue to be explored in more depth. Common links in pathogenesis that may explain the potential pathophysiological relationship between NAFLD and AH include:
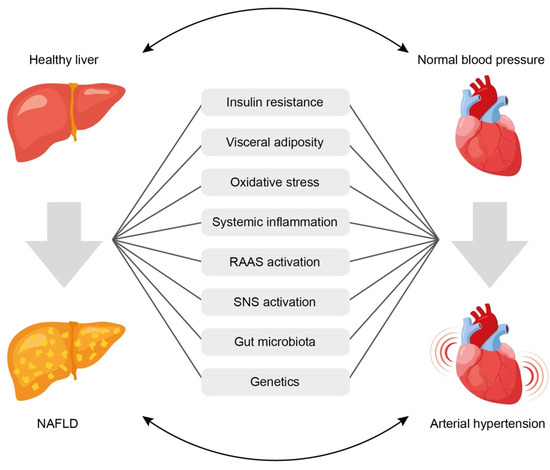
Figure 1. Common pathophysiological links underlining the association between arterial hypertension and non-alcoholic fatty liver disease. See text for explanations. NAFLD = non-alcoholic fatty liver disease; RAAS = renin-angiotensin-aldosterone system; SNS = sympathetic nervous system.
- (1)
-
Insulin resistance,
- (2)
-
Systemic inflammation,
- (3)
-
Activation of the renin-angiotensin-aldosterone system (RAAS),
- (4)
-
Oxidative stress,
- (5)
-
Activation of the sympathetic nervous system,
- (6)
-
Gut microbiota disbalance,
- (7)
-
Genetic factors.
2. Insulin Resistance
Uncontrolled AH leads to a decrease in peripheral circulation, which contributes to a reduction in the sensitivity of peripheral tissues to insulin and, consequently, to hyperinsulinemia. The latter stimulates the proliferation of smooth muscle cells and vascular fibroblasts, which leads to a narrowing of their lumen and an increase in total peripheral vascular resistance (TPVR). These vascular effects of insulin are mediated through the mitogen-activated protein kinase (MAPK) pathway, which promotes secretion of the vasoconstrictor endothelin-1 (ET-1). The other pathway that may be involved in the vascular deleterious effects of insulin is the phosphatidylinositol 3-kinase (PI3K)-dependent insulin signaling required for metabolic actions of insulin. It was shown that the impairment of PI3K/protein kinase B (Akt) pathway consistent with insulin resistance caused by glucotoxicity, lipotoxicity, or inflammation predicted diminished NO production and increased ET-1 secretion characteristic of diabetes and endothelial dysfunction [71][14]. Moreover, according to experimental data, hyperinsulinemia increases the activity of the central sections of the sympathetic nervous system, accompanied by augmented sympathetic stimulation of the heart, blood vessels, and kidneys [72][15].
There is evidence that compensatory hyperinsulinemia contributes to the activation of the RAAS and the melanocortin system of the brain, which play a pivotal role in the onset of AH [73][16]. Upon activation of the RAAS, a cascade of reactions occurs, including the production of angiotensin II, which causes spasm of smooth muscles of arterioles, an increase in hydrostatic pressure in the glomeruli, along with activation of aldosterone synthesis and higher sodium reabsorption in the kidneys. This ultimately leads to an increase in circulating blood volume and elevated BP. The kidneys are also a target organ for hyperinsulinemia. Insulin receptors are expressed on renal tubular cells and podocytes. Under physiological conditions, insulin induces vasodilation by increasing endothelial nitric oxide production through activation of the PI3K/ Akt pathway. In insulin resistance, this pathway is disrupted, and insulin triggers the MAPK pathway, which promotes renal vasoconstriction [18,72][15][17]. Besides this, under conditions of hyperinsulinemia, the activity of transmembrane ion-exchange Na+, K+ and Ca2+ ATPases is disrupted, while sodium reabsorption in the nephron tubules increases, which leads to fluid retention and the development of hypervolemia, as well as an increase in the sodium content in the blood vessel walls and their spasm [72][15].
3. Visceral Obesity
Visceral adipose tissue is an independent endocrine organ. Adipocytes secrete hormonally active molecules called adipokines (tumor necrosis factor α [TNF-α], IL-6, leptin, etc.), which enhance the pathological effects of insulin. TNF-α and IL-6 trigger the processes of cytotoxic inflammatory responses and chronic persistent inflammation both in the liver and in the vascular walls [74][18]. Adipocytes are also capable of synthesizing angiotensin II, thereby regulating BP levels [75][19].
It is known that adipokines can influence the RAAS. An increase in leptin levels against the background of a high-fat diet in rats caused the activation of the RAAS and the production of proinflammatory cytokines in the brain, which led to a progressive increase in BP [76][20]. Accordingly, L. D’Elia et al. (2021) showed that leptin values greater than 2.9 ng/mL were associated with a twofold increased risk of developing arterial stiffening in a sample of adult men without baseline arterial stiffening and antihypertensive treatment at baseline during an 8-year follow-up. These results were independent of body weight and BP [77][21]. Altered adipokine profile characterized by an increase in leptin concentration and a decrease in adiponectin levels was also described in NAFLD, thus indicating an additional common pathophysiological link between NAFLD and AH [78][22]. In addition to the effects described at the beginning of this paragraph, unfavorable action of leptin on BP level may be also attributed to the following mechanisms: reduction in NO bioavailability and regulation of ET-1 expression, as well as stimulation of endothelial cell growth and cardiovascular smooth muscle cell proliferation, increase in sympathetic nerve activity, and impairment in sodium handling [79,80][23][24].
An excess of free fatty acids (FFAs) released by adipose tissue under conditions of insulin resistance contributes to the formation of ectopic fat deposits in the form of perivascular and perirenal fat depots. In the insulin-resistant state, perivascular fat cells increase in size and number, and secrete reduced levels of regenerative factors, such as hepatocyte growth factor, and increased levels of proinflammatory factors, such as IL-6, TNF-α and monocyte chemoattractive factor 1 MCP-1. These proinflammatory factors inhibit the PI3K/Akt axis of insulin signal transduction, increase endothelial permeability and enhance insulin resistance [72][15]. This promotes the remodeling of the vascular wall with an increase in arterial stiffness and a resulting increase in pulse pressure [72,75,81][15][19][25]. Perirenal fat, along with inflicting mechanical pressure, is lipotoxic and can impair renal function in a paracrine manner, which results in RAAS activation and increased sodium reabsorption [75][19].
4. Oxidative Stress and Biologically Active Substances
Excessive release of FFAs from adipose tissue is lipotoxic to the liver. Against the background of mitochondrial dysfunction, the processes of lipid peroxidation are triggered with the formation of free radicals. An increase in free radical activity is the major process leading to the release of proinflammatory profibrogenic cytokines and hepatokines (fetuin-A, fibroblast growth factor 21 [FGF-21], and selenoprotein P) [82][26].
Some authors revealed the relationship between NAFLD and the biomarker of systemic endothelial dysfunction, E-selectin. E-selectin is a cell adhesion molecule expressed on endothelial cells; it is produced when they are damaged by cytokines. It was shown that the extent of NAFLD histological activity correlated with the level of E-selectin expression in the liver and the concentration of E-selectin in blood plasma [83][27]. Also, it was established that patients with AH and NAFLD had more pronounced endothelial function disorders, according to the values of arterial stiffness, compared with patients with isolated AH [84][28]. NAFLD is characterized by a change in the profile of hepatokines, which are biologically active substances secreted by hepatocytes. It was confirmed that they can participate in the development of systemic insulin resistance both directly (by influencing the insulin signaling pathway) and indirectly (by regulating lipid and glucose metabolism) [82][26]. For instance, in the study by T.W. Jung et al. (2013), it was demonstrated that fetuin-A might directly cause insulin resistance and modulate inflammatory reactions via stimulation of triacylglycerol accumulation in hepatocytes [85][29]. Accordingly, a significant decrease in circulating fetuin-A levels after 12 weeks of caloric restriction was accompanied by improvements in visceral fat area, blood pressure, lipid profiles, and glucose levels [86][30].
Regarding FGF-21, it was shown that circulating FGF-21 positively correlated with the brachial–ankle pulse wave velocity reflecting arterial stiffness. This suggests that FGF-21 might also be secreted by endothelial cells (not only hepatocytes) in response to stress, and that its elevated levels may be a signal of endothelial cell injury [87][31].
Regarding selenoprotein P, most published studies focus on the investigation of its role in the development and progression of pulmonary hypertension. Its effects in this condition are explained by the promotion of cell proliferation and apoptosis resistance through increased oxidative stress and mitochondrial dysfunction, which in turn are associated with activated hypoxia-inducible factor-1α and dysregulated glutathione metabolism [88,89][32][33].
The development of oxidative stress and the presence of systemic inflammation, accompanied by the circulation of TNF-α, IL-6, and advanced glycation end products, are key components of endothelial dysfunction. They can lead to both micro- and macroangiopathy and can contribute to progressive pathological remodeling of the vascular wall [81,90,91][25][34][35]. Elevated production of proinflammatory cytokines is a crucial mechanism involved in the progression of AH. TNF-α and IL-6 can regulate the expression of RAAS components, especially the production of angiotensinogen, which causes vasoconstriction, affects renal hemodynamics, and ultimately leads to an increased BP [92][36].
The presence of hepatic steatosis can negatively affect the regulation of the cardiovascular function via the autonomic nervous system. In patients with NAFLD, autonomic dysfunction was revealed in the form of an increased sympathetic activity and an impaired response to parasympathetic signals. Accordingly, the heart rate increased, and cardiac output decreased, which caused an additional impact on the cardiovascular system [93][37].
5. Gut Microbiota
An altered composition of the intestinal microbiota and a change in the permeability of the intestinal mucosa play an important role in the pathogenesis of both NAFLD and AH. The mechanisms of the gut microbiota impact on the development and progression of NAFLD and AH are caused by complex interactions between the gut microbiota and the host organism. Likely factors include metabolism of choline, bile acids, and amino acids resulting in synthesis of vasoactive hormones, such as trimethylamine (TMA) and trimethylamine N-oxide (TMAO), uremic toxins (indoxyl sulfate and p-cresol sulfate), as well as production of short-chain fatty acids (SCFAs) and ethanol by intestinal microorganisms [94,95][38][39].
5.1. Short-Chain Fatty Acids
In patients with AH and NAFLD, a reduction in the bacterial diversity of the intestinal microbiota and a decrease in the production of SCFA were revealed. The most important and biologically active SCFAs include acetate, propionate, and butyrate; they are made from dietary fiber in the intestines. Regarding their role in the pathogenesis of AH, animal studies suggested that SCFAs may have both hypotensive and hypertensive effects depending on the receptors they bind to [95,96][39][40]. For instance, propionate-induced activation of the G protein-coupled receptor (GPR) 42 in vascular endothelium caused a decrease in BP in Olfr78−/− mice [97][41]. In contrast, binding of propionate or acetate to the olfactory receptor (Olfr 78 in mice and OR51E2 in humans) increased BP, possibly due to their effect on vascular smooth muscle cells in renal afferent arterioles and peripheral blood vessels, as well as renin production [98][42]. The opposite effect of SCFAs on BP can be explained by the different sensitivity of Olfr78 and GPR41 to SCFAs. Indeed, GPR41 receptors are activated by basal serum concentrations of SCFAs ranging from 0.1 to 0.9 mmol, resulting in vasodilation and a reduction in BP. However, only higher serum levels of SCFA can activate Olfr78, thereby increasing renin production and BP values [98][42].
Moreover, by acting on GPR expressed in the sympathetic ganglia, SCFAs can directly regulate the sympathetic nervous system. They can also activate vagal afferent neurons and directly affect the function of the central nervous system. Such mechanisms of SCFA action on the central and peripheral nervous system lead to a decrease in BP [99][43].
The other mechanism of BP regulation by SCFAs includes their impact on the immune pathways, including intestinal and immune homeostasis, inflammatory cell biology and inflammatory response [99][43]. For instance, it was shown that butyrate was able to decrease the IL-6 and TNF-α levels caused by angiotensin II in vitro and induce regulatory T cells differentiation in vivo and in vitro. In addition, this SCFA could reduce IL-17 levels in patients with hypertension [100,101][44][45]. The anti-inflammatory effect of SCFAs, especially butyrate, may be also mediated by histone deacetylase inhibition in vascular endothelial cells, thereby contributing to the prevention of vascular inflammation [102][46].
Potential mechanisms of protective action of SCFAs on the liver include reduction of fat accumulation by promoting lipolysis, fatty acid oxidation, inhibition of fatty acid synthesis [103,104][47][48], maintenance of intestinal barrier function [105][49], regulation of intestinal motility [106][50], and suppression of inflammation and steatosis in the liver [107,108][51][52].
Since both NAFLD and AH are believed to be associated with metabolic disorders, the described protective effects of SCFAs, which are important for the pathogenesis of NAFLD, may also play a role in the development and progression of AH.
5.2. Trimethylamine and Trimethylamine N-oxide
Despite many studies examining the role of TMA and TMAO in the development and progression of NAFLD, there is no consensus regarding their effects on this liver disease. Some studies suggest that TMAO contributes to liver damage and disease progression [109[53][54][55][56],110,111,112], while others argue that TMA and TMAO may have a protective effect via improving lipid metabolism disorders, endoplasmic reticulum stress, and reducing cell death under lipid overload conditions [113,114,115][57][58][59]. The presented data suggest that the effect of TMAO may vary and strongly depends on the levels of other substrates in plasma, such as cholesterol [116][60]. Similarly, the role of TMA/TMAO in the development of AH remains uncertain. Some published data imply a vasoconstrictive mechanism of action of TMA/TMAO at high doses [117[61][62],118], while at low concentrations, these substances are thought to reduce diastolic dysfunction and cardiac fibrosis [119][63]. These data were confirmed by the results of meta-analysis published by X. Ge et al. (2020), indicating that, in comparison with low circulating TMAO concentrations, high TMAO concentrations were associated with a higher prevalence of hypertension (risk ratio [RR]: 1.12; 95% CI: 1.06, 1.17; p < 0.0001) [120][64].
Several potential mechanisms by which TMAO promotes hypertension include [121][65]: (1) enhanced angiotensin II-induced vasoconstriction and acute pressor response. This mechanism is implemented through the activation of the protein kinase RNA-like endoplasmic reticulum kinase (PERK) pathway, leading to apoptosis, inflammation, and vascular injury [122,123][66][67]; (2) the upregulation of scavenger receptors on the surface of macrophages and the stimulation of foam cell formation, atherosclerosis, vascular constriction and arterial stiffening [122,123][66][67]; (3) the disturbance of the reverse transport of cholesterol from extrahepatic organs and tissues into the liver resulting in increased oxidized-low density lipoprotein deposition in peripheral tissues, which contributes to atherosclerosis progression and increases the risk of CVD [124][68]; (4) increased production of proinflammatory cytokines, such as IL-1β, IL-18, TNF-α in combination with decreased production of anti-inflammatory cytokines, in particular IL-10 [123,125][67][69]; (5) participation in the development of renal dysfunction [126][70], and (6) the induction of cardiac dysfunction at high serum concentrations [127][71] by facilitating cardiac mitochondrial dysfunction, myocardial hypertrophy, and fibrosis [128][72].
5.3. Increased Intestinal Permeability
With increased intestinal permeability, gram-negative bacterial lipopolysaccharides reach the portal circulation and stimulate the development of systemic inflammation through toll-like receptors [129,130,131][73][74][75]. Moreover, it was shown that high circulating endotoxin levels resulted in the release of pro-inflammatory cytokines, including IL-1β, IL-12, IL-6, and TNF-α [132][76]. At the same time, high circulating levels of pro-inflammatory cytokines may contribute to intestinal junction disassembly and increase in gut permeability [133][77]. Indeed, an increase in BP was associated with a decrease in the level of tight junction proteins (occludin, tight junction protein 1 and claudin 4) in the rat model of AH compared with the control group [134][78]. The negative impact of arterial hypertension on gut barrier permeability may be explained by several possible mechanisms. The first one is the decreased intestinal blood flow due to changes in arterioles, particularly, thickening of the walls and narrowing of the lumens. This may lead to intestinal mucosa damage and barrier impairment. The second mechanism involves gut microbiota dysbiosis characterized by decreased microbial richness and diversity, reduced SCFAs production, and overgrowth of opportunistic pathogens [135][79].
According to the results of a meta-analysis conducted by J. Luther et al. (2015), almost 39.1% of NAFLD patients included in the analysis (n = 128) had signs of increased intestinal permeability vs. 6.8% in the control group of healthy individuals [136][80]. These data were further supported by another meta-analysis showing that impaired intestinal permeability in patients with NAFLD was associated with the grade of hepatic steatosis [137][81]. Similarly to patients with AH, it was assumed that patients with NAFLD (and especially with cirrhosis associated with NASH) had qualitative and quantitative changes in tight junction proteins [138,139][82][83]. Increased translocation of bacterial products was believed to lead to inflammation and fibrogenesis in the liver through toll-like receptor 4 stimulation. However, the relationship between intestinal permeability and fibrosis stage in NAFLD has not yet been established [140][84].
6. Genetic Factors
In addition to external factors, the role of genetic factors in the development of NAFLD and AH have been investigated. In the study of overlapping genes, 13 common genes for NAFLD and AH were identified. Besides this, four more common genes have been identified: leptin (LEP), adiponectin (ADIPOQ), aryl hydrocarbon receptor (AHR), and transforming growth factor beta-1 (TGFB1) genes, the expression of which is typical for patients with AH, NAFLD, liver fibrosis, and the presence of systemic inflammation [141][85]. In addition, C. Ma et al. (2021) showed that hypertension genes were more adjacent to NAFLD genes than random genes in the protein–protein interaction network, indicating a strong association between these two diseases [141][85].
According to expression information obtained from Gene cards database, both RNA-sequencing and microarray data indicated that RAS constituents, including a classical angiotensin-converting enzyme (ACE)/angiotensin II/type 1 angiotensin receptor axis and an alternative angiotensin-converting enzyme 2/angiotensin 1–7/Mas axis, were expressed in normal liver, heart and kidney. It is well-established that angiotensin II promotes insulin resistance, de novo lipogenesis, and pro-inflammatory cytokine production, and triggers liver inflammation and fibrogenesis [142][86], while active angiotensin 1–7 signal inhibits liver lipogenesis, fatty acid oxidation, inflammation, and fibrosis [143][87].
Another gene that seems to be related to both liver physiology and hypertension-NAFLD interaction is the aldehyde dehydrogenase (ALDH1A1, alternatively known as retinaldehyde dehydrogenase 1 or RALDH1) gene, encoding the enzyme which catalyzes the second and irreversible step of retinaldehyde oxidation to vitamin A (retinoic acid) [141][85]. According to the study by G. Zhong et al. (2019), the expression of ALDH1A1 was significantly higher in the livers of NASH patients than in the livers of healthy volunteers [144][88]. Moreover, analysis of the Oncomine database demonstrated that the expression of ALDH1A1 was significantly upregulated in the hepatocellular carcinoma tissues than in the normal tissues [145][89]. It is considered that ALDH1A1 might play an important role in the detoxification of lipid-derived aldehydes, including 4-hydroxy-2-nonenal and acrolein, which are known for their ability to mediate oxidative stress [146][90]. Additionally, through binding to either retinoic acid receptors (RARs) or retinoid X receptors (RXRs), retinoic acid may exert opposite effects on lipid metabolism in the liver: RARs binding leads to the suppression of hepatic non-esterified free fatty acids and triglyceride accumulation, while RXR-mediated signaling may cause hepatic lipid accumulation [147][91].
It was also revealed that polymorphism of the angiotensin II receptor type 1 (AGTR1) gene was indicative of the occurrence of NAFLD and AH in patients without obesity, insulin resistance, or hyperlipidemia. Some studies demonstrated that carriers of the C allele of the AGTR1 gene exhibited an elevated release of FFAs from adipose tissue and an imbalance of adipokines with a predominance of proinflammatory adipokines and chemokines over anti-inflammatory adiponectin [148][92].
Understanding the shared genes and biological mechanisms of AH and NAFLD can help to develop combined preventive strategies, explore novel therapeutic approaches against NAFLD, and design the most suitable treatment plan for patients with comorbidity of AH and NAFLD.
7. Influence of Gender on the Development of AH and NAFLD
It is well known that the frequency and prevalence of AH and NAFLD depend on the gender and age of the patient.
Most current guidelines for the management of AH patients agree that its prevalence among men and women of different age groups is different [149,150][93][94]. This may be due to various biological and physiological factors, as well as their interaction [151][95]. Similar to NAFLD, the prevalence of AH in the age group under 50 years is significantly higher in men than in women, while after reaching 50 years of age, the prevalence of AH in women is approximately the same as it is in men, or even slightly higher [149,150,152][93][94][96]. The likely biological factors determining the unequal prevalence of AH in men and women are the effects of sex hormones and differences in the sex chromosomes [153][97], while probable behavioral factors are high values of body mass index [154][98], smoking [155][99] and low physical activity [156][100].
A twelve-year Japanese study established that the prevalence of fatty liver in men averaged 26% vs. 13% in women. Also, it was shown that the frequency of NAFLD in women increases with age to a greater extent than in men, and the maximum differences were recorded in the age group of 70–79 years. On the contrary, the incidence of this pathology in men was similar in all age groups [157,158,159][101][102][103]. Another study from South China reported that the prevalence of NAFLD in the age group under 50 years old was significantly higher in men compared with women (22.4% vs. 7.1%, respectively, p < 0.001). However, this pattern was reversed in the age group of 50 years and older (20.6% vs. 27.6%, respectively, p < 0.05) [158,160][102][104].
There is strong evidence suggesting an important role of sex hormones in the onset of NAFLD. A meta-analysis showed that an increase in serum testosterone levels in women was also observed in polycystic ovary syndrome (PCOS), which is one of the conditions associated with NAFLD; this was accompanied by an increased risk of developing NAFLD in women. Hypogonadism associated with PCOS was shown to double the risk of developing NAFLD and contribute to an increase in obesity, insulin resistance, and metabolic syndrome [161,162,163][105][106][107]. Meanwhile, a decrease in serum testosterone levels is associated with the risk of hepatic steatosis in men [164][108]. However, a few studies suggested some protective effect of androgens regarding the development of hepatic steatosis. They can promote exocytosis of very-low-density lipoproteins (VLDL), inhibition of de novo lipogenesis, maintenance of carbohydrate metabolism, and fatty acid beta-oxidation [165,166][109][110].
Estrogens play a protective role against the development of hepatic steatosis in both men and women. This effect is implemented due to the ability of estrogens to increase insulin sensitivity, reduce the synthesis of triglycerides, and activate free fatty acid oxidation in the liver, as well as improve mitochondrial function and reduce the severity of inflammation in the liver tissue [165,167,168,169][109][111][112][113]. Estrogens are also involved in maintaining cholesterol metabolism in the liver by means of ensuring the synthesis of lipoproteins, secretion of VLDL, increased formation of high-density lipoproteins (HDL) and elimination of oxidized low-density lipoproteins (LDL).
In addition to the well-known role of sex hormones in the development of metabolic syndrome and related disorders, some studies elucidated that sex chromosomes per se may be responsible for the sexual dimorphism seen in NAFLD and metabolic syndrome. Sex chromosomes independent of gonadal status in a mouse model were associated with simple steatosis and NAFLD [170][114]. A study by Chen et al. (2016) demonstrated that the sex chromosome complement, XX or XY, affected adiposity and weight gain, because XX mice exhibited higher adiposity than XY mice even after a gonadectomy was performed to eliminate the effect of sex hormones [171][115]. It has been argued that increased adiposity was due to the presence of an extra X chromosome rather than the absence of a Y chromosome, as shown by genetic studies in mice with XO and XXY chromosomes. The established mechanism involved certain genes avoiding X chromosome inactivation and exhibiting increased levels of expression in the adipose tissue and liver of XX mice rather than XY mice [158,171][102][115].
With regard to the combination of NAFLD and AH, the following age- and gender-related characteristics have been described. First, women with NAFLD had a higher prevalence of essential hypertension (75.8%) [172][116]. Second, women with AH were more likely to develop NAFLD within 5 years [34][117]. Female gender is associated with a greater likelihood of NAFLD progression with the development of advanced liver fibrosis [173][118], especially at the age of over 50 years, which can lead to the development of pathogenetic mechanisms of AH, viz., an increase in arterial wall stiffness and endothelial dysfunction [174][119].
Gender-based differences in the prevalence of AH appear multifactorial as well, and their causes are not fully understood. In recent years, differences in the activity of the sympathetic nervous system and arterial wall stiffness were widely discussed, in the genesis of which sex hormones may play a certain role [175][120]. It should be noted that dysfunction of the autonomic nervous system may be more important for the development of AH in women than in men [176][121]. Furthermore, the age-related increase in autonomic nervous system activity is more pronounced in women than in men and does not depend on body mass index or menopause [177,178][122][123]. It is well known that the risk and incidence of AH in premenopausal women is lower than in men, but after 65 years of age, the prevalence of AH becomes higher in women [179][124].
Androgens and estrogens are able to regulate BP by influencing the RAAS. Androgens stimulate the RAAS, which leads to an elevated BP [180][125], while ovarian hormones have the opposite effect via reducing the activity of renin and ACE in blood plasma [181][126]. The effect of sex hormones on renal sodium reabsorption and vascular resistance may also explain differences between men and women in BP control [180,182][125][127]. Estrogens are likely to have the ability to maintain normal endothelial function by stimulating nitric oxide (NO) production including positive effects on arterial wall structure and function, which, in turn, help reduce vascular wall stiffness. Moreover, they reduce the effects of the sympathetic nervous system [179,183][124][128].
References
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Association of non-alcoholic fatty liver disease with major adverse cardiovascular events: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 33386.
- Kuang, M.; Lu, S.; Xie, Q.; Peng, N.; He, S.; Yu, C.; Qiu, J.; Sheng, G.; Zou, Y. Abdominal obesity phenotypes are associated with the risk of developing non-alcoholic fatty liver disease: Insights from the general population. BMC Gastroenterol. 2022, 1, 311.
- Huh, Y.; Cho, Y.J.; Nam, G.E. Recent Epidemiology and Risk Factors of Nonalcoholic Fatty Liver Disease. J. Obes. Metab. Syndr. 2022, 1, 17–27.
- Zarghamravanbakhsh, P.; Frenkel, M.; Poretsky, L. Metabolic causes and consequences of nonalcoholic fatty liver disease (NAFLD). Metabol. Open. 2021, 12, 100149, Erratum in Metabol. Open. 2023, 17, 100231.
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 7, 921–937.
- Kim, K.; Kang, K.; Sheol, H.; Shin, J.; Sim, Y.; Yang, T.; Hwang, J.; Lee, J.-M. The Association between Serum Uric Acid Levels and 10-Year Cardiovascular Disease Risk in Non-Alcoholic Fatty Liver Disease Patients. Int. J. Environ. Res. Public Health 2022, 19, 1042.
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298.
- Iglesias Morcillo, M.; Freuer, D.; Peters, A.; Heier, M.; Teupser, D.; Meisinger, C.; Linseisen, J. Association between fatty liver index and blood coagulation markers: A population-based study. Lipids Health Dis. 2023, 1, 83.
- Emamat, H.; Tangestani, H.; Behrad Nasab, M.; Ghalandari, H.; Hekmatdoost, A. The association between epicardial adipose tissue and non-alcoholic fatty liver disease: A systematic review of existing human studies. EXCLI J. 2021, 20, 1096–1105.
- Vu, H.; Khanh Tuong, T.T.; Hoang Lan, N.; Quoc Thang, T.; Bilgin, K.; Hoa, T.; Minh Duc, N.; The Dung, B. Association between nonalcoholic fatty liver disease and carotid intima-media thickness. Clin. Ter. 2023, 174, 42–47.
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2009, 2, 231–237.
- Fuchs, F.D.; Whelton, P.K. High Blood Pressure and Cardiovascular Disease. Hypertension 2020, 75, 285–292.
- Zhao, Y.C.; Zhao, G.J.; Chen, Z.; She, Z.G.; Cai, J.; Li, H. Nonalcoholic fatty liver disease: An emerging driver of hypertension. Hypertension 2020, 75, 275–284.
- Muniyappa, R.; Chen, H.; Montagnani, M.; Sherman, A.; Quon, M.J. Endothelial dysfunction due to selective insulin resistance in vascular endothelium: Insights from mechanistic modeling. Am. J. Physiol. Endocrinol. Metab. 2020, 3, E629–E646.
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Häring, H.U. The impact of insulin resistance on the kidney and vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737.
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of hyperinsulinemia and insulin resistance in hypertension: Metabolic syndrome revisited. Can. J. Cardiol. 2020, 36, 671–682.
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352.
- Oikonomou, D.; Georgiopoulos, G.; Katsi, V.; Kourek, C.; Tsioufis, C.; Alexopoulou, A.; Koutli, E.; Tousoulis, D. Non-alcoholic fatty liver disease and hypertension: Coprevalent or correlated? Eur. J. Gastroenterol. Hepatol. 2018, 30, 979–985.
- Xue, B.; Yu, Y.; Zhang, Z.; Guo, F.; Beltz, T.G.; Thunhorst, R.L.; Felder, R.B.; Johnson, A.K. Leptin mediates high-fat diet sensitization of angiotensin II-elicited hypertension by upregulating the brain renin-angiotensin system and inflammation. Hypertension 2016, 67, 970–976.
- Catena, C.; Bernardi, S.; Sabato, N.; Grillo, A.; Ermani, M.; Sechi, L.A.; Fabris, B.; Carretta, R.; Fallo, F. Ambulatory arterial stiffness indices and non-alcoholic fatty liver disease in essential hypertension. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 389–393.
- D’Elia, L.; Giaquinto, A.; Iacone, R.; Russo, O.; Strazzullo, P.; Galletti, F. Serum leptin is associated with increased pulse pressure and the development of arterial stiffening in adult men: Results of an eight-year follow-up study. Hypertens. Res. 2021, 11, 1444–1450.
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell Physiol. 2019, 12, 21630–21641.
- Poetsch, M.S.; Strano, A.; Guan, K. Role of Leptin in Cardiovascular Diseases. Front. Endocrinol. 2020, 11, 354.
- D’Elia, L.; Strazzullo, P. Excess Body Weight, Insulin Resistance and Isolated Systolic Hypertension: Potential Pathophysiological Links. High Blood Press. Cardiovasc. Prev. 2018, 1, 17–23.
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64, 924–928.
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520.
- Guzik, T.J.; Touyz, R.M. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension 2017, 70, 660–667.
- Statsenko, M.E.; Streltsova, A.M.; Turovets, M.I. Effect of non-alcoholic fatty liver disease on arterial stiffness and the risk of cardiovascular complications in patients with arterial hypertension. Russ. Arch. Intern. Med. 2020, 10, 296–304.
- Jung, T.W.; Youn, B.S.; Choi, H.Y.; Lee, S.Y.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Kim, B.H.; Baik, S.H.; Choi, K.M. Salsalate and adiponectin ameliorate hepatic steatosis by inhibition of the hepatokine fetuin-A. Biochem. Pharmacol. 2013, 7, 960–969.
- Choi, K.M.; Han, K.A.; Ahn, H.J.; Lee, S.Y.; Hwang, S.Y.; Kim, B.H.; Hong, H.C.; Choi, H.Y.; Yang, S.J.; Yoo, H.J.; et al. The effects of caloric restriction on fetuin-A and cardiovascular risk factors in rats and humans: A randomized controlled trial. Clin. Endocrinol. 2013, 3, 356–363.
- Yang, S.J.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Cho, G.J.; Hwang, T.G.; Baik, S.H.; Choi, D.S.; Kim, S.M.; Choi, K.M. Effects of a three-month combined exercise programme on fibroblast growth factor 21 and fetuin-A levels and arterial stiffness in obese women. Clin. Endocrinol. 2011, 4, 464–469.
- Kikuchi, N.; Satoh, K.; Kurosawa, R.; Yaoita, N.; Elias-Al-Mamun, M.; Siddique, M.A.H.; Omura, J.; Satoh, T.; Nogi, M.; Sunamura, S.; et al. Selenoprotein P Promotes the Development of Pulmonary Arterial Hypertension: Possible Novel Therapeutic Target. Circulation 2018, 6, 600–623.
- Sun, Q.; Hackler, J.; Hilger, J.; Gluschke, H.; Muric, A.; Simmons, S.; Schomburg, L.; Siegert, E. Selenium and Copper as Biomarkers for Pulmonary Arterial Hypertension in Systemic Sclerosis. Nutrients 2020, 12, 1894.
- Nosalski, R.; McGinnigle, E.; Siedlinski, M.; Guzik, T.J. Novel immune mechanisms in hypertension and cardiovascular risk. Curr. Cardiovasc. Risk Rep. 2017, 11, 12.
- Simons, N.; Bijnen, M.; Wouters, K.A.M.; Rensen, S.S.; Beulens, J.W.J.; van Greevenbroek, M.M.J.; ’t Hart, L.M.; Greve, J.W.M.; van der Kallen, C.J.H.; Schaper, N.C.; et al. The endothelial function biomarker soluble E-selectin is associated with nonalcoholic fatty liver disease. Liver Int. 2020, 40, 1079–1088.
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr. Hypertens. Rep. 2018, 20, 100.
- Houghton, D.; Zalewski, P.; Hallsworth, K.; Cassidy, S.; Thoma, C.; Avery, L.; Slomko, J.; Hardy, T.; Burt, A.D.; Tiniakos, D.; et al. The degree of hepatic steatosis associates with impaired cardiac and autonomic function. J. Hepatol. 2019, 70, 1203–1213.
- Sharpton, S.R.; Ajmera, V.; Loomba, R. Emerging role of the gut microbiome in nonalcoholic fatty liver disease: From composition to function. Clin. Gastroenterol. Hepatol. 2019, 17, 296–306.
- Tokarek, J.; Budny, E.; Saar, M.; Kućmierz, J.; Młynarska, E.; Rysz, J.; Franczyk, B. Does the Composition of Gut Microbiota Affect Hypertension? Molecular Mechanisms Involved in Increasing Blood Pressure. Int. J. Mol. Sci. 2023, 24, 1377.
- Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients 2020, 12, 2982.
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834.
- Miyamoto, J.; Kasubuchi, M.; Nakajima, A.; Irie, J.; Itoh, H.; Kimura, I. The role of short-chain fatty acid on blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2016, 25, 379–383.
- Wu, Y.; Xu, H.; Tu, X.; Gao, Z. The Role of Short-Chain Fatty Acids of Gut Microbiota Origin in Hypertension. Front. Microbiol. 2021, 12, 730809.
- Wang, L.; Zhu, Q.; Lu, A.; Liu, X.; Zhang, L.; Xu, C.; Liu, X.; Li, H.; Yang, T. Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin-angiotensin system. J. Hypertens. 2017, 9, 1899–1908.
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 6, 701–718.
- Chun, P. Therapeutic effects of histone deacetylase inhibitors on heart disease. Arch. Pharm. Res. 2020, 12, 1276–1296.
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50.
- Liu, L.; Fu, C.; Li, F. Acetate Affects the Process of Lipid Metabolism in Rabbit Liver, Skeletal Muscle and Adipose Tissue. Animals 2019, 9, 799.
- Xie, L.; Alam, M.J.; Marques, F.Z.; Mackay, C.R. A major mechanism for immunomodulation: Dietary fibres and acid metabolites. Semin. Immunol. 2023, 66, 101737.
- Mishima, Y.; Ishihara, S. Enteric Microbiota-Mediated Serotonergic Signaling in Pathogenesis of Irritable Bowel Syndrome. Int. J. Mol. Sci. 2021, 22, 10235.
- Skelly, A.N.; Sato, Y.; Kearney, S.; Honda, K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol. 2019, 19, 305–323.
- Chen, M.; Hui, S.; Lang, H.; Zhou, M.; Zhang, Y.; Kang, C.; Zeng, X.; Zhang, Q.; Yi, L.; Mi, M. SIRT3 Deficiency Promotes High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease in Correlation with Impaired Intestinal Permeability through Gut Microbial Dysbiosis. Mol. Nutr. Food Res. 2019, 63, 1800612.
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; de Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971.
- Aragonès, G.; Colom-Pellicer, M.; Aguilar, C.; Guiu-Jurado, E.; Martínez, S.; Sabench, F.; Antonio Porras, J.; Riesco, D.; Del Castillo, D.; Richart, C.; et al. Circulating microbiota-derived metabolites: A “liquid biopsy?”. Int. J. Obes. 2020, 44, 875–885.
- Cordeiro, A.; Costa, R.; Andrade, N.; Silva, C.; Canabrava, N.; Pena, M.J.; Rodrigues, I.; Andrade, S.; Ramalho, A. Does adipose tissue inflammation drive the development of non-alcoholic fatty liver disease in obesity? Clin. Res. Hepatol. Gastroenterol. 2020, 44, 394–402.
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, 1900257.
- Zhao, Z.H.; Xin, F.Z.; Zhou, D.; Xue, Y.Q.; Liu, X.L.; Yang, R.X.; Pan, Q.; Fan, J.G. Trimethylamine N-oxide attenuates high-fat high-cholesterol diet-induced steatohepatitis by reducing hepatic cholesterol overload in rats. World. J. Gastroenterol. 2019, 25, 2450–2462.
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585.
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 7341, 57–63.
- Vu, V.; Kim, Y.; Cho, M. Effects of SCFAs and TMAO on non-alcoholic fatty liver disease indicating the therapeutic benefits of plant-based diet, and supplemental prebiotics, probiotics and synbiotics. Appl. Biol. Chem. 2023, 66, 11.
- Maksymiuk, K.M.; Szudzik, M.; Gawryś-Kopczyńska, M.; Onyszkiewicz, M.; Samborowska, E.; Mogilnicka, I.; Ufnal, M. Trimethylamine, a gut bacteria metabolite and air pollutant, increases blood pressure and markers of kidney damage including proteinuria and KIM-1 in rats. J. Transl. Med. 2022, 20, 470.
- Brunt, V.E.; Casso, A.G.; Gioscia-Ryan, R.A.; Sapinsley, Z.J.; Ziemba, B.P.; Clayton, Z.S.; Bazzoni, A.E.; VanDongen, N.S.; Richey, J.J.; Hutton, D.A.; et al. Gut Microbiome-Derived Metabolite Trimethylamine N-Oxide Induces Aortic Stiffening and Increases Systolic Blood Pressure With Aging in Mice and Humans. Hypertension 2021, 78, 499–511.
- Huc, T.; Drapala, A.; Gawrys, M.; Konop, M.; Bielinska, K.; Zaorska, E.; Samborowska, E.; Wyczalkowska-Tomasik, A.; Pączek, L.; Dadlez, M.; et al. Chronic, low-dose TMAO treatment reduces diastolic dysfunction and heart fibrosis in hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, 1805–1820.
- Ge, X.; Zheng, L.; Zhuang, R.; Yu, P.; Xu, Z.; Liu, G.; Xi, X.; Zhou, X.; Fan, H. The Gut Microbial Metabolite Trimethylamine N-Oxide and Hypertension Risk: A Systematic Review and Dose-Response Meta-analysis. Adv. Nutr. 2020, 1, 66–76.
- Mutengo, K.H.; Masenga, S.K.; Mweemba, A.; Mutale, W.; Kirabo, A. Gut microbiota dependant trimethylamine N-oxide and hypertension. Front. Physiol. 2023, 14, 1075641.
- Jiang, S.; Shui, Y.; Cui, Y.; Tang, C.; Wang, X.; Qiu, X.; Hu, W.; Fei, L.; Li, Y.; Zhang, S.; et al. Gut microbiota dependent trimethylamine N-oxide aggravates angiotensin II-induced hypertension. Redox Biol. 2021, 46, 102115.
- Liu, Y.; Dai, M. Trimethylamine N-Oxide Generated by the Gut Microbiota Is Associated with Vascular Inflammation: New Insights into Atherosclerosis. Mediat. Inflamm. 2020, 2020, 4634172.
- Wang, B.; Qiu, J.; Lian, J.; Yang, X.; Zhou, J. Gut Metabolite Trimethylamine-N-Oxide in Atherosclerosis: From Mechanism to Therapy. Front. Cardiovasc. Med. 2021, 8, 723886.
- Huang, Y.; Lin, F.; Tang, R.; Bao, C.; Zhou, Q.; Ye, K.; Shen, Y.; Liu, C.; Hong, C.; Yang, K.; et al. Gut Microbial Metabolite Trimethylamine N-Oxide Aggravates Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2022, 4, 452–460.
- Zhou, J.; Wang, D.; Li, B.; Li, X.; Lai, X.; Lei, S.; Li, N.; Zhang, X. Relationship between Plasma Trimethylamine N-Oxide Levels and Renal Dysfunction in Patients with Hypertension. Kidney Blood Press. Res. 2021, 4, 421–432.
- Naghipour, S.; Cox, A.J.; Peart, J.N.; Du Toit, E.F.; Headrick, J.P. Trimethylamine N-oxide: Heart of the microbiota-CVD nexus? Nutr. Res. Rev. 2021, 1, 125–146.
- Li, Z.; Wu, Z.; Yan, J.; Liu, H.; Liu, Q.; Deng, Y.; Ou, C.; Chen, M. Gut microbe-derived metabolite trimethylamine N-oxide induces cardiac hypertrophy and fibrosis. Lab. Investig. 2019, 3, 346–357.
- Ma, J.; Li, H. The role of gut microbiota in atherosclerosis and hypertension. Front. Pharmacol. 2018, 9, 1082.
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340.
- Li, F.; Ye, J.; Shao, C.; Zhong, B. Compositional alterations of gut microbiota in nonalcoholic fatty liver disease patients: A systematic review and meta-analysis. Lipids Health Dis. 2021, 20, 22.
- Ntlahla, E.E.; Mfengu, M.M.; Engwa, G.A.; Nkeh-Chungag, B.N.; Sewani-Rusike, C.R. Gut permeability is associated with hypertension and measures of obesity but not with Endothelial Dysfunction in South African youth. Afr. Health Sci. 2021, 3, 1172–1184.
- Sturgeon, C.; Fasano, A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 2016, 4, e1251384.
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-linked pathophysiological alterations in the gut. Circ. Res. 2017, 120, 312–323.
- Li, C.; Xiao, P.; Lin, D.; Zhong, H.J.; Zhang, R.; Zhao, Z.G.; He, X.X. Risk Factors for Intestinal Barrier Impairment in Patients With Essential Hypertension. Front. Med. 2021, 7, 543698.
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232.
- De Munck, T.J.I.; Xu, P.; Verwijs, H.J.A.; Masclee, A.A.M.; Jonkers, D.; Verbeek, J.; Koek, G.H. Intestinal permeability in human nonalcoholic fatty liver disease: A systematic review and meta-analysis. Liver Int. 2020, 40, 2906–2916.
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887.
- Muñoz, L.; Caparrós, E.; Albillos, A.; Francés, R. The shaping of gut immunity in cirrhosis. Front. Immunol. 2023, 14, 1139554.
- Forlano, R.; Mullish, B.H.; Roberts, L.A.; Thursz, M.R.; Manousou, P. The Intestinal Barrier and Its Dysfunction in Patients with Metabolic Diseases and Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 662.
- Ma, C.; Yan, K.; Wang, Z.; Zhang, Q.; Gao, L.; Xu, T.; Sai, J.; Cheng, F.; Du, Y. The association between hypertension and nonalcoholic fatty liver disease (NAFLD): Literature evidence and systems biology analysis. Bioengineered 2021, 12, 2187–2202.
- Roderburg, C.; Krieg, S.; Krieg, A.; Demir, M.; Luedde, T.; Kostev, K.; Loosen, S.H. Non-alcoholic fatty liver disease (NAFLD) is associated with an increased incidence of chronic kidney disease (CKD). Eur. J. Med. Res. 2023, 1, 153.
- Hartl, L.; Rumpf, B.; Domenig, O.; Simbrunner, B.; Paternostro, R.; Jachs, M.; Poglitsch, M.; Marculescu, R.; Trauner, M.; Reindl-Schwaighofer, R.; et al. The systemic and hepatic alternative renin-angiotensin system is activated in liver cirrhosis, linked to endothelial dysfunction and inflammation. Sci. Rep. 2023, 1, 953.
- Zhong, G.; Kirkwood, J.; Won, K.J.; Tjota, N.; Jeong, H.; Isoherranen, N. Characterization of Vitamin A Metabolome in Human Livers With and Without Nonalcoholic Fatty Liver Disease. J. Pharmacol. Exp. Ther. 2019, 1, 92–103.
- Yang, C.K.; Wang, X.K.; Liao, X.W.; Han, C.Y.; Yu, T.D.; Qin, W.; Zhu, G.Z.; Su, H.; Yu, L.; Liu, X.G.; et al. Aldehyde dehydrogenase 1 (ALDH1) isoform expression and potential clinical implications in hepatocellular carcinoma. PLoS ONE 2017, 8, e0182208.
- Makia, N.L.; Bojang, P.; Falkner, K.C.; Conklin, D.J.; Prough, R.A. Murine hepatic aldehyde dehydrogenase 1a1 is a major contributor to oxidation of aldehydes formed by lipid peroxidation. Chem. Biol. Interact. 2011, 1–3, 278–287.
- Saeed, A.; Dullaart, R.P.F.; Schreuder, T.C.M.A.; Blokzijl, H.; Faber, K.N. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2017, 1, 29.
- Musso, G.; Saba, F.; Cassader, M.; Paschetta, E.; De Michieli, F.; Pinach, S.; Framarin, L.; Berrutti, M.; Leone, N.; Parente, R.; et al. Angiotensin II type 1 receptor rs5186 gene variant predicts incident NAFLD and associated hypertension: Role of dietary fat-induced pro-inflammatory cell activation. Am. J. Gastroenterol. 2019, 114, 607–619.
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104, Erratum in Eur. Heart J. 2019, 40, 475.
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 13–115, Erratum in Hypertension 2018, 71, 140–144.
- Connelly, P.J.; Currie, G.; Delles, C. Sex Differences in the Prevalence, Outcomes and Management of Hypertension. Curr. Hypertens. Rep. 2022, 6, 185–192.
- Meinert, F.; Thomopoulos, C.; Kreutz, R. Sex and gender in hypertension guidelines. J. Hum. Hypertens. 2023, 8, 654–661.
- Sandberg, K.; Ji, H. Sex differences in primary hypertension. Biol. Sex. Differ. 2012, 3, 7.
- Khalid, F.; Siddique, A.; Siddiqui, J.A.; Panhwar, G.; Singh, S.; Anwar, A.; Hashmi, A.A. Correlation Between Body Mass Index and Blood Pressure Levels Among Hypertensive Patients: A Gender-Based Comparison. Cureus 2020, 12, e10974.
- Vallée, A. Associations between smoking and alcohol consumption with blood pressure in a middle-aged population. Tob. Induc. Dis. 2023, 21, 61.
- Egan, B.M. Physical Activity and Hypertension: Knowing Is Not Enough; We Must Apply. Willing Is Not Enough; We Must Do-von Goethe. Hypertension 2017, 69, 404–406.
- Kojima, S.; Watanabe, N.; Numata, M.; Ogawa, T.; Matsuzaki, S. Increase in the prevalence of fatty liver in Japan over the past 12 years: Analysis of clinical background. J. Gastroenterol. 2003, 38, 954–961.
- Nagral, A.; Bangar, M.; Menezes, S.; Bhatia, S.; Butt, N.; Ghosh, J.; Manchanayake, J.H.; Mahtab, M.A.; Singh, S.P. Gender Differences in Nonalcoholic Fatty Liver Disease. Euroasian J. Hepatogastroenterol. 2022, 12 (Suppl. S1), S19–S25.
- Martin-Grau, M.; Monleon, D. Sex dimorphism and metabolic profiles in management of metabolic-associated fatty liver disease. World J. Clin. Cases. 2023, 11, 1236–1244.
- Zhou, Y.J.; Li, Y.Y.; Nie, Y.Q.; Ma, J.X.; Lu, L.G.; Shi, S.L.; Chen, M.H.; Hu, P.J. Prevalence of fatty liver disease and its risk factors in the population of South China. World J. Gastroenterol. 2007, 13, 6419–6424.
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in endocrine diseases. Endocr. Connect. 2021, 10, R52–R65.
- Singeap, A.M.; Stanciu, C.; Huiban, L.; Muzica, C.M.; Cuciureanu, T.; Girleanu, I.; Chiriac, S.; Zenovia, S.; Nastasa, R.; Sfarti, C.; et al. Association between Nonalcoholic Fatty Liver Disease and Endocrinopathies: Clinical Implications. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6678142.
- Chen, M.J.; Ho, H.N. Hepatic manifestations of women with polycystic ovary syndrome. Best. Pract. Res. Clin. Obstet. Gynaecol. 2016, 37, 119–128.
- Jaruvongvanich, V.; Sanguankeo, A.; Riangwiwat, T.; Upala, S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 382–394.
- Yang, M.; Ma, F.; Guan, M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 320.
- Della Torre, S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells 2021, 10, 2502.
- Varlamov, O.; Bethea, C.L.; Roberts, C.T., Jr. Sex-specific differences in lipid and glucose metabolism. Front. Endocrinol. 2015, 5, 241.
- Ceccarelli, I.; Bioletti, L.; Peparini, S.; Solomita, E.; Ricci, C.; Casini, I.; Miceli, E.; Aloisi, A.M. Estrogens and phytoestrogens in body functions. Neurosci. Biobehav. Rev. 2022, 132, 648–663.
- Von-Hafe, M.; Borges-Canha, M.; Vale, C.; Leite, A.R.; Sérgio Neves, J.; Carvalho, D.; Leite-Moreira, A. Nonalcoholic Fatty Liver Disease and Endocrine Axes-A Scoping Review. Metabolites 2022, 12, 298.
- Link, J.C.; Chen, X.; Arnold, A.P.; Reue, K. Metabolic impact of sex chromosomes. Adipocyte 2013, 2, 74–79.
- Chen, X.; McClusky, R.; Chen, J.; Beaven, S.W.; Tontonoz, P.; Arnold, A.P.; Reue, K. The number of x chromosomes causes sex differences in adiposity in mice. PLoS Genet. 2012, 8, e1002709.
- Michopoulos, S.; Chouzouri, V.I.; Manios, E.D.; Grapsa, E.; Antoniou, Z.; Papadimitriou, C.A.; Zakopoulos, N.; Dimopoulos, A.M. Untreated newly diagnosed essential hypertension is associated with nonalcoholic fatty liver disease in a population of a hypertensive center. Clin. Exp. Gastroenterol. 2016, 9, 1–9.
- Liu, P.; Tang, Y.; Guo, X.; Zhu, X.; He, M.; Yuan, J.; Wang, Y.; Wei, S.; Chen, W.; Zhang, X.; et al. Bidirectional association between nonalcoholic fatty liver disease and hypertension from the Dongfeng-Tongji cohort study. J. Am. Soc. Hypertens. 2018, 12, 660–670.
- Balakrishnan, M.; Patel, P.; Dunn-Valadez, S.; Dao, C.; Khan, V.; Ali, H.; El-Serag, L.; Hernaez, R.; Sisson, A.; Thrift, A.P.; et al. Women Have a Lower Risk of Nonalcoholic Fatty Liver Disease but a Higher Risk of Progression vs Men: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2021, 19, 61–71.
- Villela-Nogueira, C.A.; Leite, N.C.; Cardoso, C.R.; Salles, G.F. NAFLD and Increased Aortic Stiffness: Parallel or Common Physiopathological Mechanisms? Int. J. Mol. Sci. 2016, 17, 460.
- Cadeddu, C.; Franconi, F.; Cassisa, L.; Campesi, I.; Pepe, A.; Cugusi, L.; Maffei, S.; Gallina, S.; Sciomer, S.; Mercuro, G.; et al. Arterial hypertension in the female world: Pathophysiology and therapy. J. Cardiovasc. Med. 2016, 17, 229–236.
- Sevre, K.; Lefrandt, J.D.; Nordby, G.; Os, I.; Mulder, M.; Gans, R.O.; Rostrup, M.; Smit, A.J. Autonomic function in hypertensive and normotensive subjects: The importance of gender. Hypertension 2001, 37, 1351–1356.
- Narkiewicz, K.; Phillips, B.G.; Kato, M.; Hering, D.; Bieniaszewski, L.; Somers, V.K. Gender-selective interaction between aging, blood pressure, and sympathetic nerve activity. Hypertension 2005, 45, 522–525.
- Matsukawa, T.; Sugiyama, Y.; Watanabe, T.; Kobayashi, F.; Mano, T. Gender difference in age-related changes in muscle sympathetic nerve activity in healthy subjects. Am. J. Physiol. 1998, 275, R1600–R1604.
- Meloni, A.; Cadeddu, C.; Cugusi, L.; Donataccio, M.P.; Deidda, M.; Sciomer, S.; Gallina, S.; Vassalle, C.; Moscucci, F.; Mercuro, G.; et al. Gender Differences and Cardiometabolic Risk: The Importance of the Risk Factors. Int. J. Mol. Sci. 2023, 24, 1588.
- Reckelhoff, J.F. Gender differences in the regulation of blood pressure. Hypertension 2001, 37, 1199–1208.
- Oparil, S.; Miller, A.P. Gender and blood pressure. J. Clin. Hypertens. 2005, 7, 300–309.
- Barris, C.T.; Faulkner, J.L.; Belin de Chantemèle, E.J. Salt Sensitivity of Blood Pressure in Women. Hypertension 2023, 80, 268–278.
- Mercuro, G.; Podda, A.; Pitzalis, L.; Zoncu, S.; Mascia, M.; Melis, G.B.; Rosano, G.M. Evidence of a role of endogenous estrogen in the modulation of autonomic nervous system. Am. J. Cardiol. 2000, 85, 787–789.
More