Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Satoru Matsuda and Version 2 by Peter Tang.
Cisplatin-induced acute kidney injury (AKI) is the main factor restraining the clinical application of cisplatin. As increased levels of reactive oxygen species (ROS) may promote the progression of the injury, the elimination of ROS has been considered as an effective method to prevent the cisplatin-induced AKI. In addition, it has been revealed that an inducer of autophagy could protect kidney cells in the autophagy dependent manner. Induction of autophagy could also modulate the production of ROS in cases of renal injury.
- cisplatin
- reactive oxygen species
- autophagy
- histone deacetylases
- cancer
- acute kidney injury
1. Introduction
Cisplatin is one of the most widely used broad-spectrum anticancer agents, and is used for the treatment of various solid tumors such as ovarian cancer, prostate cancer, bladder cancer, head or neck cancer, and lung cancer [1][2][1,2]. The antitumor mechanisms of cisplatin are mainly DNA damage via the enhanced generation of reactive oxygen species (ROS) [3]. The excess ROS such as superoxide anion, hydrogen peroxide, and hydroxyl radical would cause oxidative damage to various important molecules including proteins, lipids, and/or DNAs, leading to the critical damage of cancer cells [4]. It had been suggested that hydrogen peroxide is involved in the cisplatin-induced necrosis, whereas hydroxyl radical is responsible for the cisplatin-induced apoptosis [5]. Accordingly, the protective effects of hydroxyl radical scavengers are associated with an inhibition of cytochrome c release and caspase activation [5]. In general, cancer cells have higher levels of ROS than normal cells as a result of hyper-metabolism [6]. In addition to their extreme cytotoxicity, cisplatin could also have a variety of non-specific adverse reactions in cancer patients [7]. Studies have suggested that the accumulation of intracellular ROS is a hallmark of cisplatin-induced acute kidney injury (AKI) [8]. Cisplatin may show high activity in the fast proliferating cells, thereby causing cellular damage. In particular, about 30% of cisplatin-administered patients suffer from renal dysfunction and/or injury [9]. Increased formation of ROS in renal proximal convoluted tubule cells may be associated with the cisplatin-induced AKI [10], which is a substantial complication of cisplatin chemotherapy related to the ROS-dependent death of renal cells [11]. The AKI may be associated with high morbidity and mortality. To date, there are few strategies for preventing cisplatin-induced AKI [12], and it is urgent to search for novel therapeutic procedures to protect the kidney against the nephrotoxicity of cisplatin. In recent years, remarkable advances have been made in effective protective regimens for the nephrotoxicity of cisplatin [13]. Basically, protection of kidney from cisplatin-induced AKI might be attainable with antioxidant-based therapeutic interventions that increase antioxidant levels and thus improve the damage from ROS.
2. ROS and Autophagy in Cisplatin-Induced Acute Kidney Injury
The excessive generation of ROS has been regarded as the critical role during the pathogenetic process induced by cisplatin, by which DNA damage and/or cell death could occur. Increased ROS production is also known to change the mitochondrial electron transport chain, and eventually lead to apoptosis [14][19]. In particular, the dysfunction of the mitochondrial respiratory chain results in the further excess production of ROS that contributes to severe kidney injury [15][20]. Consequently, the mitochondrial dysfunction induced by the treatment with cisplatin could be considered as due to increased levels of ROS resulting in various cellular apoptosis including kidney cells [2][16][2,21]. Increased levels of ROS can also contribute to the tubular cell apoptosis in kidney, thereby causing more severe kidney injury during AKI [17][22]. Further generation of ROS in the tubular cells mitochondria might thus possibly contribute to the exacerbation of cisplatin-induced AKI. Therefore, the elimination of ROS has long been considered as an effective procedure to prevent the cisplatin-induced AKI [18][23]. In addition to increased ROS levels, treatment with cisplatin also impairs the activity of antioxidants such as superoxide dismutases (SODs), catalase, and glutathione peroxidase, which could function to reduce ROS levels [19][24]. The SODs are ROS-eliminating super enzymes with several subcellular localizations [20][25]. Targeting the cisplatin-induced oxidative stress via manipulation of the cellular antioxidant system including the expression of SODs could be beneficial for protecting against cisplatin nephrotoxicity. In fact, some agents with antioxidant and/or anti-inflammation activities could alleviate the cisplatin-induced cell damage by reduced production of ROS [21][26]. These therapeutic approaches may enhance the tolerance to cisplatin and hence might enable greater dose intensity associated with better outcomes. It has been shown that sirt1 expression on proximal tubules in the kidney may save cisplatin-induced AKI by preserving the function of peroxisomes and the elimination of ROS, which could be a potential therapeutic target for the treatment of cisplatin-induced AKI [22][27] (Figure 1).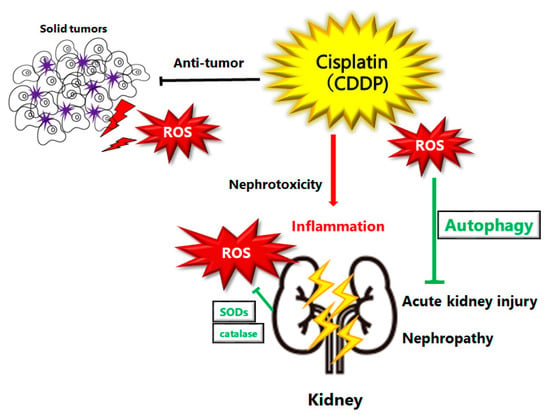
Figure 1. Schematic illustration of pathogenesis of cisplatin induced acute kidney injury or nephropathy. Reactive oxygen species (ROS), inflammation, and autophagy are all involved in the pathogenesis of cisplatin induced acute kidney injury. ROS may damage DNA or organelles within a cell. The damage could be treated with autophagy to enhance the survival of kidney cells. If the damage is too severe to be repaired, cells might undergo cell-death leading to kidney injury or nephropathy. Note that several significant features have been omitted for clarity.