Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Sirius Huang and Version 2 by Sirius Huang.
Primary amenorrhea (PA) describes the complete absence of menses by the age of 15 years. It is a devastating diagnosis that can affect the adolescent’s view of her femininity, sexuality, fertility and self-image. A normal menstrual cycle can occur only in the presence of: a properly functioning hypothalamus–pituitary axis, well-developed and active ovaries, outflow tract without abnormalities. Any dysfunction in any of these players can result in amenorrhea.
- amenorrhea
- adolescents
- primary ovarian insufficiency
- hypogonadotropic hypogonadism
- turner syndrome
1. Introduction
Primary amenorrhea (PA) describes the complete absence of menses by the age of 15 years. It is a devastating diagnosis that can affect the adolescent’s view of her femininity, sexuality, fertility and self-image [1].
The downward secular trend in breast development onset is well-known and contrasts with the small reduction in the median age of menarche. In an epidemiological study that included 4250 school-age girls, researchers found that menarche occurred at a median age of 12.5 years and that 95% of girls had reached menarche by the age of 14 years [2]. Pubertal menstrual disorders, such as heavy menstrual bleeding, are commonly observed in the first 2 years after menarche. On the other hand, prolonged amenorrhea beyond the age of 14 years is not normal and needs to be evaluated.
The menstrual cycle is considered a “vital sign”. Therefore, PA is an important clinical indicator that must be investigated.
A normal menstrual cycle can occur only in the presence of:
- -
-
A properly functioning hypothalamus–pituitary axis;
- -
-
Well-developed and active ovaries;
- -
-
Outflow tract without abnormalities.
PA concerns approximately 0.1% of girls [4]. Despite this low prevalence, PA is often the presenting sign of an underlying endocrine disease, chronic disease, gene alteration, or Müllerian duct anomalies.
PA in adolescence is likely to require multi-disciplinary management (pediatrician endocrinologist, gynecologist, surgeon, psychologist, and fertility team) and a sensitive, age-appropriated approach that takes into account the adolescents’ emotional maturity.
The diagnosis must be promptly confirmed. When necessary, estrogen replacement therapy should be proposed for pubertal development and psychological improvement [5].
2. Definition
PA can be seen as an opportunity for the early diagnosis/treatment of conditions that affect the hypothalamus–pituitary–ovarian axis [6]. It may be associated with significant morbidity and can allow for the early identification and management of health issues with potential negative consequences for adult life [7]. Indeed, the menstrual cycle should be considered as a “vital” sign.
PA is a risk factor of early and late consequences, in function of the degree of estrogenization. In adolescents with high estrogen levels, peri-pubertal hyperestrogenism constitutes a risk of endometrium hyperplasia that causes dysfunctional uterine bleeding. Later in life, it is a risk of endometrial cancer and breast cancer. Conversely, in estrogen-deficient adolescents, a reduction in bone mineral density increases the life-long risk of bone fractures [8] and of cardiovascular diseases [9]. In addition, psychological problems and even psychiatric disorders should not be overlooked.
There is no consensus on which PA type requires investigations. PA management should be started in four conditions:
- -
-
Adolescent who did not reach menarche by the age of 14 years;
- -
-
Adolescent who did not reach menarche after more than 3 years since thelarche occurrence;
- -
-
Adolescent who did not reach menarche by the age of 13 years and without secondary sexual characteristic development;
- -
-
Adolescent who did not reach menarche by the age 14 years and with suspected eating disorder or excessive exercise, with signs of hyperandrogenism, or with failure to thrive.
3. Causes of Primary Amenorrhea
According to the American Society for Reproductive Medicine (ASRM) practice committee [10], PA can be associated with anatomic defects of the outflow tract, primary hypogonadism (XX, X0, XY), hypothalamic causes (dysfunction, Kallman syndrome, chronic illness), pituitary disorders (prolactinoma, infection diseases), other endocrine gland disorders (adrenal, thyroid, ovary), or multifactorial causes (polycystic ovary syndrome, PCOS). This classification does not reflect routine practice. Researchers prefer to consider that PA could be related to endocrine defects (hypothalamus–pituitary–ovarian axis alterations), genetic abnormalities, previous radiotherapy or/and chemotherapy, metabolic diseases, autoimmune disorders, infections, exposure to endocrine-disrupting chemicals (EDCs), Müllerian tract defects, or unknown factors.
In recent decades, PA was mainly explained by outflow tract anatomic defects (10%), ovarian causes (30%), pituitary causes (5%), hypothalamic causes (10%), functional causes (30%), and unknown causes (30–35%).
In the literature, PA is mainly caused by (Table 1):
- 1—Endocrine defects of the hypothalamus–pituitary–ovarian axis.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 2—Müllerian defects (Normogonadotropic amenorrhea) |
- 2—Genetic defects of the ovary.
- 3—Metabolic diseases.
- 4—Autoimmune diseases.
- 5—Infections.
- 6—Iatrogenic causes (radiotherapy, chemotherapy).
- 7—Müllerian tract defects.
- 8—Environmental factors.
- 9—Idiopathic
Table 1. The main causes of adolescent primary amenorrhea.
| 1—Endocrine Defects within the Hypothalamo–Pituitary–Ovarian Axis (HPOa) |
|
|
|
|
|
|
|
|
|
|
|
3.1. Endocrine Defects of the Hypothalamus–Pituitary–Ovarian Axis
When puberty does not occur at the extreme end of the normal spectrum (i.e., constitutional delay of growth and puberty), hypogonadism must be suspected. There are two types of hypogonadism: hypogonadotropic hypogonadism and hypergonadotropic hypogonadism. Hypogonadotropic hypogonadism can be transient due to an underlying medical condition, or persistent due to a gonadotropin-releasing hormone (GnRH) defect. Midline congenital defects, such as cleft lip and palate, and neural tube defects are suggestive of permanent hypogonadotropic hypogonadism. Hypergonadotropic hypogonadism is due to gonad failure.
3.1.1. Hypogonadotropic Hypogonadism
Hypogonadotropic hypogonadism (CHH) may be congenital and due to isolated FSH-LH deficiency or multiple gonadotropin deficiencies. It may be secondary to an acquired hypothalamic–pituitary disease. Hypothalamic–pituitary diseases include Kallmann syndrome, Prader–Willi syndrome, congenital hypopituitarism, and septo-optic dysplasia, among others.
Congenital Hypogonadotropic Hypogonadism
Hypogonadotropic hypogonadism may be due to gene alterations, and their identification facilitates a differential diagnosis and then management:
- -
-
The genes encoding kisspeptin and its receptor (KISS1 and KISS1R) and neurokinin B and its receptor (TAC3 and TACR3), which regulate GnRH release, should be the first to be screened in clinical settings for equivocal cases, such as delayed puberty versus idiopathic hypogonadotropic hypogonadism, because they are the main causes of GnRH pulse generator defects.
- -
-
In Kallmann syndrome, the screening of specific genes should be prioritized based on their association with clinical features: synkinesis (KAL1), dental agenesis (FGF8/FGFR1), bone anomalies (FGF8/FGFR1), and hearing loss (CHD7, SOX1). New genes have been recently identified and the list of genes involved in hypogonadotropic hypogonadism is still growing.
More than 25 different genes have been implicated in congenital hypogonadotropic hypogonadism and Kallmann syndrome, which account for ~50% of CHH cases.
Acquired Hypogonadotropic Hypogonadism
The absence of maturation of the hypothalamic–pituitary–ovarian axis may be secondary to acquired hypothalamic–pituitary diseases, such as:
- -
-
Brain tumors: craniopharyngioma, astrocytoma;
- -
-
Central nervous system infiltration diseases (e.g., histiocytosis);
- -
-
Chemo- or radiotherapy;
- -
Functional Hypothalamic Amenorrhea
The diagnosis of functional hypothalamic amenorrhea (FHA) is carried out after having excluded all anatomic and organic causes of PA [12]. According to Gordon et al., 2017, FHA is often associated with stress, weight loss, excessive exercise, and their combination [13].
The evaluation of adolescents with suspected FHA includes a detailed history focused on diet, eating disorders, exercise, athletic training, perfectionism, and ambition.
3.1.2. Hypergonadotropic Hypogonadism
Hypergonadotropic hypogonadism can be congenital or acquired. In function of the karyotype, it can classified in:
- -
-
XX hypergonadotropic hypogonadism;
- -
-
X0 hypergonadotropic hypogonadism;
- -
-
XY hypergonadotropic hypogonadism.
XX Hypergonadotropic Hypogonadism
- (a)
-
Premature ovarian insufficiency
Premature ovarian insufficiency (POI) ranges from 1 in 100 to 1 in 10,000 for women aged younger than 40 years with increasing prevalence at each decade of life and is specifically uncommon in adolescents. POI is characterized by severe estrogen deficiency due to a loss of ovarian function, which can be congenital or acquired [14]. The prevalence of known gene alterations that may be linked to PA is estimated at ~20%. To date, 18 POI-causing genes have been identified: BMP15, DMC1, EIF2S2, FIGLA, FOXL2, FSHR, GDF9, GPR3, HFM1, LHX8, MSH5, NOBOX, NR5A1, PGRMC1, STAG3, XPNPEP2, BHLB, and FSHB.
In a recent analysis of these genes by next-generation sequencing in patients with PA, Eskenazi et al. showed that 25% of these adolescents presented at least one variant, and 18% presented a variant of unknown significance. In this study, NOBOX variants were the most frequently detected (19% of all patients) [15]. Ghosh et al. confirmed that chromosomal anomalies are one of the major causes of amenorrhea in India [16]. BMP15 mutations and FMR1 pre-mutation (fragile X syndrome) have been detected in ~10% of adolescents with PA [15].
- (b)
-
Metabolic disorders
-
Classic galactosemia affects around 1/25,000 of new-born girls and is due to mutations in the GALT gene that decrease/abolish galactose-1-phosphate uridylyltranserase activity, leading to the toxic accumulation of galactose in the ovaries and in the whole body. Several mechanisms have been postulated, including the direct toxicity of galactose to oocytes and follicles, leading to accelerated atresia of the ovarian pool [17].
-
Thalassemia and sickle cell disease are the most prevalent inherited hemoglobin disorders (recessive pattern). Transfusion-related iron overload may lead to gonad dysfunction, the absence of puberty development, and PA [18].
-
Other metabolic disorders also may be associated with PA [19], such as congenital adrenal hyperplasia (due to 17-hydroxylase enzyme deficiency) and aromatase deficiency. In some girls, PA may be associated with type 1 diabetes, low BMI and abnormal pulsatile GnRH secretion.
-
Obesity: adolescents with severe obesity usually have elevated levels of plasma androgens. The normalization of plasma androgens upon weight loss leads to the resumption of ovulation, suggesting that overweight-related hyperandrogenism is the cause of amenorrhea in adolescent girls with obesity [20].
-
PCOS: It is not rare that PA may lead to the detection of PCOS, especially when severe insulin resistance is present in the peri-pubertal period in adolescents with central obesity, early hyperinsulinism and insulin resistance [21,22][21][22]. Many PCOS features appear during early adolescence, such as oligomenorrhea, heavy menstrual bleeding, and signs of hyperandrogenism. PA is an uncommon manifestation of PCOS (1.4 to 14% of girls present PA as an initial feature of PCOS) [21]. Early hyperinsulinism, insulin resistance, and central obesity are observed in girls with severe PCOS [22].
-
- (c)
-
Autoimmune diseases
Autoimmune diseases (diabetes mellitus, hypothyroidism, Hashimoto thyroiditis, Grave disease) are common disorders associated with PA [23].
Evidence for an autoimmune mechanism involved in POI is based on the description of lymphocytic oophoritis, associated with autoantibodies against ovarian antigens [24]. PA is also associated with other autoimmune disorders of the adrenal gland, thyroid, and pancreas. Autoimmune polyglandular syndrome type 1, caused by AIRE gene mutations, is also associated with PA. Autoimmune disorders can also be associated with non-endocrine diseases, such as candidiasis, vitiligo, systemic lupus, rheumatoid arthritis.
- (d)
-
Infections
Young women living with HIV are 70% more likely to experience amenorrhea.
The overall prevalence of amenorrhea among women with HIV is ~5%. In a recent meta-analysis, King et al. showed a significant association between HIV and amenorrhea (OR = 1.68) [25]. It is unclear whether amenorrhea might be a complication of HIV infection or of other risk factors, such as low BMI, wasting (collapse of GnRH secretion), opiate and anti-psychotic use (anovulation), immunosuppression and chemotherapy. According to Cejtin et al., amenorrhea is reversible in 37% of women with HIV reporting this problem [26].
- (e)
-
Iatrogenic causes (radiotherapy, chemotherapy)
Modern pediatric cancer management is becoming highly effective, and therefore, it is crucial to monitor the endocrine and gynecological consequences in adolescents who underwent such treatments [27,28][27][28]. It is thought that in this population, the prevalence of ovarian failure is ~10%, although Jablonska et al. found that 31.6% of cancer survivors experienced amenorrhea [29], and Chemaitilly et al. reported ovarian insufficiency rates from 2.1 to 92.2% [30]. In the ovarian follicles, both oocytes and granulosa cells are vulnerable to chemotherapy effects. In addition, damage to blood vessels and focal fibrosis of the ovarian cortex are involved in chemotherapy-induced ovarian damage [31].
Besides chemotherapy with alkylating agents, abdominal/pelvic radiation must be considered a risk factor. In adolescents who received total body irradiation before stem cell transplant, AMH, which is considered a good marker of the ovarian reserve [32], was undetectable in serum [33]. Oocytes are very sensitive to radiotherapy. The detrimental effects are influenced by the irradiation field, dose and fractionation schedules. AMH levels are lower in children who received radiotherapy to the abdomen, pelvis and total body for cancer management [34].
- (f)
-
Environmental factors (lifestyle, EDCs)
Many EDCs target the ovary, with effects particularly on folliculogenesis and steroidogenesis during fetal life [36]. Several experimental data demonstrated EDC harmful effects on follicle growth by promoting atresia [37]. Moreover, some EDCs (i.e., pesticides, phthalates, bisphenol-A, and dioxin) alter ovarian steroidogenesis. Severe acute and chronic prenatal exposure to EDCs may impair ovarian function later in life and disturb puberty [38].
- (g)
-
Idiopathic
In the past, not informing young patients about the diagnosis and treatment was the standard practice. It is now established practice to gradually disclose the diagnosis and the underlying cause to adolescents, in function of their level of understanding and knowledge. This leads to better adherence to the treatment and allows for the screening of other family members, if relevant.
X0 Hypergonadotropic Hypogonadism: Turner Syndrome
Turner syndrome is the most common example of hypergonadotropic hypogonadism and explains up to 30% of all PA. It is associated with X chromosome numerical or structural alterations. Its prevalence is ~50/100,000 girls in Caucasian populations. Although 20% of patients with Turner syndrome begin puberty spontaneously, only very few progress to menarche. The absence of puberty and PA are very frequent clinical manifestations of Turner syndrome [39]. Ovarian dysgenesis and early follicular apoptosis are key features of this syndrome, resulting in POI with estrogen deficiency in the peri-pubertal period [40]. AMH plasma concentration is a useful marker of ovarian function.
XY Hypergonadotropic Hypogonadism: Disorders of Sex Development (DSD)
The term 46, XY DSD is used to describe 46, XY adolescents with undermasculinization, leading in some cases to a female phenotype. In Denmark, the prevalence of 46, XY females was estimated to be 6.4 per 100,000 live born females [41]. Testosterone, AMH, FSH and LH serum concentration and the presence of Müllerian derivatives on pelvic ultrasound are used to differentiate gonad dysgenesis (associated with the insufficient gonadal secretion of testosterone and AMH) from androgen production defects or androgen resistance (Figure 1).
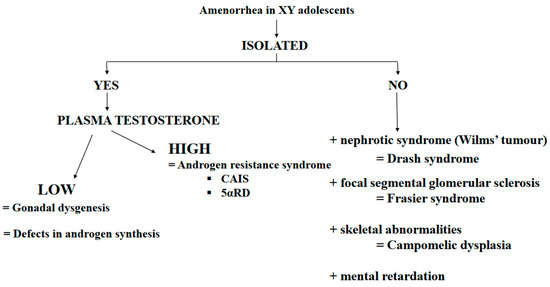
Figure 1. Main causes of amenorrhea in XY adolescents. Abbreviation: CAIS = complete androgen insensitivity syndrome; 5αRD = 5α-reductase deficiency.
- (a)
-
Gonad dysgenesis:
Gonad dysgenesis is a genetic defect in testis determination characterized by variable alterations of Leydig and Sertoli cell function. This disorder can be secondary to mutations in any of the several genes implicated in the primitive gonad differentiation to testes.
SRY gene alterations lead to 46, XY sex reversal with a female phenotype. The diagnosis of Swyer syndrome is made at puberty in girls with PA [42].
In some cases, gonad dysgenesis has been linked to mutations of SF1, a gene involved in the development of male gonads and adrenal glands.
In some patients, gonad dysgenesis is associated with kidney dysfunction. In these patients, the diagnosis of Drash syndrome (i.e., Wilms’ tumor and kidney insufficiency) or Frasier syndrome (i.e., proteinuria secondary to focal glomerular sclerosis) may be made. Both syndromes are due to disease-specific WT1 gene abnormalities. Indeed, heterozygous mutations in the open reading frame have been associated with Drash syndrome, while intron mutations leading to splicing abnormalities have been found in patients with Frasier syndrome.
Several SOX9 mutations have been identified in patients with severe skeletal malformations (e.g., campomelic dysplasia) associated with sex reversal and gonad dysgenesis.
Homozygous or composite heterozygous mutations of the desert hedgehog (DHH) gene, which is implicated in testis differentiation and perineal development, have been identified. These patients present a female phenotype, sometimes with neuropathy.
- (b)
-
Testosterone Production Defects
Defects in testosterone production are rare and are characterized by severe external genital undervirilization. Conversely, no Müllerian derivatives are present because AMH is normally secreted by Sertoli cells. These defects are due to an enzymatic defect in testosterone biosynthesis or are secondary to alterations in the gene encoding the LH receptor.
Leydig cell agenesis (or hypoplasia) is a rare form of 46, XY DSD. The typical presentation is PA, no breast development at puberty, and low testosterone at baseline and after human chorionic gonadotropin (hCG) stimulation. This condition is determined by a homozygous or double heterozygous inactivating mutation of the LH receptor gene.
Defects in 3-β-hydroxysteroid dehydrogenase are associated with variable, but insufficient virilization in 46, XY boys, ranging from a female phenotype to minor DSD forms.
Similarly, 17-α-hydroxylase defects lead to a variable phenotype. In some patients, the diagnosis is made only at puberty due to PA. An excess of 11-deoxycorticosterone due to CYP17 gene alterations (recessive transmission) causes hypertension during puberty.
Defects of 17-β-hydroxysteroid reductase are rare, but cause testicular block and deficits in testosterone production, leading frequently to a female phenotype.
- (c)
-
Androgen-resistance disorders
Androgen-resistance disorders are characterized by normal/high testosterone and AMH production. These disorders are caused by an androgen receptor (AR) defect or 5α-reductase deficiency.
-
Complete androgen insensitivity syndrome (CAIS)
CAIS is often diagnosed during puberty in girls with PA, normal breast development, and sparse axillary and pubic hair [43]. The endocrine work-up shows high plasma testosterone and LH concentrations. The diagnosis is confirmed by the identification of an AR gene mutation.
-
5α-reductase deficiency
The phenotype is typically female, but all degrees of undervirilization can be observed [44,45][44][45]. The diagnosis is usually made at puberty because of PA, the absence of breast development, and striking virilization (hirsutism, clitoral hypertrophy, significant muscle development, and behavior masculinization). The diagnosis is confirmed by identifying a mutation in the gene encoding 5α-reductase 2.
3.2. Normogonadotropin Ovulatory Amenorrhea: Congenital Müllerian Defects
Müllerian anomalies concern 4 to 5% of women [46]. Several classifications have been proposed, but without a consensus. The most used and accepted was proposed by the ASRM and classifies these anomalies in 12 classes [47].
Congenital malformations of the female genital organs include the absence of a uterus and vagina, and some obstructive abnormalities of the reproductive tract [48]. Müllerian aplasia or hypoplasia, also known as Mayer–Rokitanski–Kuster–Hauster syndrome, may be isolated or associated with other congenital malformations [49].
Transverse vaginal septum is caused by the persistence of the vaginal plate after it meets the Müllerian tract. Examination reveals a shortened, blind vaginal pouch.
An imperforated hymen usually presents as a bluish bulging mass due to hematocolpos at the vagina entrance.
References
- Klein, D.A.; Paradise, S.L.; Reeder, R.M. Amenorrhea: A Systematic Approach to Diagnosis and Management. Am. Fam. Physician 2019, 100, 39–48.
- Sultan, C.; Gaspari, L.; Maimoun, L.; Kalfa, N.; Paris, F. Disorders of puberty. Best. Pract. Res. Clin. Obstet. Gynaecol. 2018, 48, 62–89.
- Golden, N.H.; Carlson, J.L. The Pathophysiology of Amenorrhea in the Adolescent. Ann. N. Y. Acad. Sci. 2008, 1135, 163–178.
- Timmreck, L.S.; Reindollar, R.H. Contemporary issues in primary amenorrhea. Obstet. Gynecol. Clin. N. Am. 2003, 30, 287–302.
- Master-Hunter, T.; Heiman, D.L. Amenorrhea: Evaluation and treatment. Am. Fam. Physician 2006, 73, 1374–1382.
- Popat, V.B.; Prodanov, T.; Calis, K.A.; Nelson, L.M. The Menstrual Cycle: A biological marker of general health in adolescents. Ann. N. Y. Acad. Sci. 2008, 1135, 43–51.
- American Academy of Pediatrics; Committee on Adolescence; American College of Obstetricians and Gynecologists; Committee on Adolescent Health Care. Menstruation in Girls and Adolescents: Using the Menstrual Cycle as a Vital Sign. Pediatrics 2006, 118, 2245–2250.
- Gordon, C.M.; Nelson, L.M. Amenorrhea and bone health in adolescents and young women. Curr. Opin. Obstet. Gynecol. 2003, 15, 377–384.
- Simoncini, T.; Genazzani, A.R.; Giannini, A. The Long-Term Cardiovascular Risks Associated with Amenorrhea. In Frontiers in Gynecological Endocrinology; Sultan, C., Genazzani, A., Eds.; ISGE Series; Springer: Cham, Switzerland, 2017.
- Practice Committee of American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil. Steril. 2008, 90, S219–S225.
- Fazeli, P.; Nachtigall, L.B. Hyperprolactinemia and Pituitary Causes of Amenorrhea. In Amenorrhea; Santoro, N., Neal-Perry, G., Eds.; Contemporary Endocrinology; Humana Press: Totowa, NJ, USA, 2010.
- Pedreira, C.C.; Maya, J.; Misra, M. Functional hypothalamic amenorrhea: Impact on bone and neuropsychiatric outcomes. Front. Endocrinol. 2022, 13, 953180.
- Gordon, C.M.; Ackerman, K.E.; Berga, S.L.; Kaplan, J.R.; Mastorakos, G.; Misra, M.; Murad, M.H.; Santoro, N.F.; Warren, M.P. Functional Hypothalamic Amenorrhea: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2017, 102, 1413–1439.
- Kanner, L.; Hakim, J.C.; Kankanamge, C.D.; Patel, V.; Yu, V.; Podany, E.; Gomez-Lobo, V. Noncytotoxic-Related Primary Ovarian Insufficiency in Adolescents: Multicenter Case Series and Review. J. Pediatr. Adolesc. Gynecol. 2018, 31, 597–604.
- Eskenazi, S.; Bachelot, A.; Hugon-Rodin, J.; Plu-Bureau, G.; Gompel, A.; Catteau-Jonard, S.; Molina-Gomes, D.; Dewailly, D.; Dodé, C.; Christin-Maitre, S.; et al. Next Generation Sequencing Should Be Proposed to Every Woman with “Idiopathic” Primary Ovarian Insufficiency. J. Endocr. Soc. 2021, 5, bvab032.
- Ghosh, S.; Roy, S.; Halder, A. Study of frequency and types of chromosomal abnormalities in phenotypically female patients with amenorrhea in Eastern Indian population. J. Obstet. Gynaecol. Res. 2020, 46, 1627–1638.
- van Erven, B.; Berry, G.T.; Cassiman, D.; Connolly, G.; Forga, M.; Gautschi, M.; Gubbels, C.S.; Hollak, C.E.; Janssen, M.C.; Knerr, I.; et al. Fertility in adult women with classic galactosemia and primary ovarian insufficiency. Fertil. Steril. 2017, 108, 168–174.
- Mamsen, L.S.; Kristensen, S.G.; Pors, S.E.; Bøtkjær, J.A.; Ernst, E.; Macklon, K.T.; Gook, D.; Kumar, A.; Kalra, B.; Andersen, C.Y. Consequences of β-Thalassemia or Sickle Cell Disease for Ovarian Follicle Number and Morphology in Girls Who Had Ovarian Tissue Cryopreserved. Front. Endocrinol. 2021, 11, 593718.
- Yoon, J.Y.; Cheon, C.K. Evaluation and management of amenorrhea related to congenital sex hormonal disorders. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 149–157.
- Itriyeva, K. The effects of obesity on the menstrual cycle. Curr. Probl. Pediatr. Adolesc. Health Care 2022, 52, 101241.
- Rachmiel, M.; Kives, S.; Atenafu, E.; Hamilton, J. Primary Amenorrhea as a Manifestation of Polycystic Ovarian Syndrome in Adolescents: A unique subgroup? Arch. Pediatr. Adolesc. Med. 2008, 162, 521–525.
- Ledger, W.L.; Skull, J. Amenorrhoea: Investigation and treatment. Curr. Obstet. Gynaecol. 2004, 14, 254–260.
- Szeliga, A.; Calik-Ksepka, A.; Maciejewska-Jeske, M.; Grymowicz, M.; Smolarczyk, K.; Kostrzak, A.; Smolarczyk, R.; Rudnicka, E.; Meczekalski, B. Autoimmune Diseases in Patients with Premature Ovarian Insufficiency—Our Current State of Knowledge. Int. J. Mol. Sci. 2021, 22, 2594.
- Komorowska, B. Autoimmune premature ovarian failure. Menopause Rev. 2016, 15, 210–214.
- King, E.M.; Albert, A.; Murray, M.C. HIV and amenorrhea: A meta-analysis. AIDS 2019, 33, 483–491.
- Cejtin, H.E.; Evans, C.T.; Greenblatt, R.; Minkoff, H.; Weber, K.M.; Wright, R.; Colie, C.; Golub, E.; Massad, L.S. Prolonged Amenorrhea and Resumption of Menses in Women with HIV. J. Women’s Health 2018, 27, 1441–1448.
- van der Kooi, A.-L.L.F.; Mulder, R.L.; Hudson, M.M.; Kremer, L.C.; Skinner, R.; Constine, L.S.; van Dorp, W.; Broeder, E.V.D.-D.; Falck-Winther, J.; Wallace, W.H.; et al. Counseling and surveillance of obstetrical risks for female childhood, adolescent, and young adult cancer survivors: Recommendations from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Am. J. Obstet. Gynecol. 2020, 224, 3–15.
- Beneventi, F.; Locatelli, E.; Giorgiani, G.; Zecca, M.; Locatelli, F.; Cavagnoli, C.; Simonetta, M.; Bariselli, S.; Negri, B.; Spinillo, A. Gonadal and uterine function in female survivors treated by chemotherapy, radiotherapy, and/or bone marrow transplantation for childhood malignant and non-malignant diseases. BJOG Int. J. Obstet. Gynaecol. 2014, 121, 856–865; discussion 865.
- Jablonska, O.; Shi, Z.; Valdez, K.E.; Ting, A.Y.; Petroff, B.K. Temporal and anatomical sensitivities to the aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin leading to premature acyclicity with age in rats. Int. J. Androl. 2010, 33, 405–412.
- Chemaitilly, W.; Li, Z.; Krasin, M.J.; Brooke, R.J.; Wilson, C.L.; Green, D.M.; Klosky, J.L.; Barnes, N.; Clark, K.L.; Farr, J.B.; et al. Premature Ovarian Insufficiency in Childhood Cancer Survivors: A Report from the St. Jude Lifetime Cohort. J. Clin. Endocrinol. Metab. 2017, 102, 2242–2250.
- Gargus, E.; Deans, R.; Anazodo, A.; Woodruff, T.K. Management of Primary Ovarian Insufficiency Symptoms in Survivors of Childhood and Adolescent Cancer. J. Natl. Compr. Cancer Netw. 2018, 16, 1137–1149.
- Fong, S.L.; Laven, J.; Hakvoort-Cammel, F.; Schipper, I.; Visser, J.; Themmen, A.; de Jong, F.; Heuvel-Eibrink, M.v.D. Assessment of ovarian reserve in adult childhood cancer survivors using anti-Mullerian hormone. Hum. Reprod. 2008, 24, 982–990.
- Wong, Q.H.Y.; Anderson, R.A. The role of antimullerian hormone in assessing ovarian damage from chemotherapy, radiotherapy and surgery. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 391–398.
- Kim, H.-A.; Choi, J.; Park, C.S.; Seong, M.-K.; Hong, S.-E.; Kim, J.-S.; Park, I.-C.; Lee, J.K.; Noh, W.C.; ASTRRA Trial Investigators. Post-chemotherapy serum anti-Müllerian hormone level predicts ovarian function recovery. Endocr. Connect. 2018, 7, 949–956.
- Reifschneider, K.; Auble, B.A.; Rose, S.R. Update of Endocrine Dysfunction following Pediatric Traumatic Brain Injury. J. Clin. Med. 2015, 4, 1536–1560.
- Patel, S.; Zhou, C.; Rattan, S.; Flaws, J.A. Effects of Endocrine-Disrupting Chemicals on the Ovary. Biol. Reprod. 2015, 93, 20.
- Vabre, P.; Gatimel, N.; Moreau, J.; Gayrard, V.; Picard-Hagen, N.; Parinaud, J.; Léandri, R. Environmental pollutants, a possible etiology for premature ovarian insufficiency: A narrative review of animal and human data. Environ. Health 2017, 16, 37.
- Gigante, E.; Picciocchi, E.; Valenzano, A.; Dell’Orco, S. The effects of the endocrine disruptors and of the halogens on the female reproductive system and on epigenetics: A brief review. Acta Medica Mediterr. 2018, 34, 1295.
- Hoag, B.D.; Tsai, S.L.; Williams, D.D.; Cernich, J.T. International Guideline Adherence in Girls with Turner Syndrome: Multiple Subspecialty Clinics versus Coordinated Multidisciplinary Clinic. Endocr. Pract. 2022, 28, 1203–1209.
- Trolle, C.; Mortensen, K.; Hjerrild, B.E.; Cleemann, L.; Gravholt, C.H. Clinical care of adult Turner syndrome—New aspects. Pediatr. Endocrinol. Rev. 2012, 9 (Suppl. 2), 739–749.
- Berglund, A.; Johannsen, T.H.; Stochholm, K.; Viuff, M.H.; Fedder, J.; Main, K.M.; Gravholt, C.H. Incidence, Prevalence, Diagnostic Delay, and Clinical Presentation of Female 46,XY Disorders of Sex Development. J. Clin. Endocrinol. Metab. 2016, 101, 4532–4540.
- Meyer, K.F.; Filho, L.G.F.; Silva, K.I.; Trauzcinsky, P.A.; Reuter, C.; Souza, M.B.M. The XY female and SWYER syndrome. Urol. Case Rep. 2019, 26, 100939.
- Sultan, C.; Lumbroso, S.; Paris, F.; Jeandel, C.; Terouanne, B.; Belon, C.; Audran, F.; Poujol, N.; Georget, V.; Gobinet, J.; et al. Disorders of Androgen Action. Semin. Reprod. Med. 2002, 20, 217–228.
- Maimoun, L.; Philibert, P.; Cammas, B.; Audran, F.; Bouchard, P.; Fenichel, P.; Cartigny, M.; Pienkowski, C.; Polak, M.; Skordis, N.; et al. Phenotypical, Biological, and Molecular Heterogeneity of 5α-Reductase Deficiency: An Extensive International Experience of 55 Patients. J. Clin. Endocrinol. Metab. 2011, 96, 296–307.
- Maimoun, L.; Philibert, P.; Bouchard, P.; Öcal, G.; Leheup, B.; Fenichel, P.; Servant, N.; Paris, F.; Sultan, C. Primary amenorrhea in four adolescents revealed 5α-reductase deficiency confirmed by molecular analysis. Fertil. Steril. 2011, 95(804.e1–804.e5).
- Deligeoroglou, E.; Athanasopoulos, N.; Tsimaris, P.; Dimopoulos, K.D.; Vrachnis, N.; Creatsas, G. Evaluation and management of adolescent amenorrhea. Ann. N. Y. Acad. Sci. 2010, 1205, 23–32.
- Passos, I.d.M.P.e.; Britto, R.L. Diagnosis and treatment of müllerian malformations. Taiwan J. Obstet. Gynecol. 2020, 59, 183–188.
- Kapczuk, K.; Kędzia, W. Primary Amenorrhea Due to Anatomical Abnormalities of the Reproductive Tract: Molecular Insight. Int. J. Mol. Sci. 2021, 22, 11495.
- Sultan, C.; Biason-Lauber, A.; Philibert, P. Mayer–Rokitansky–Kuster–Hauser syndrome: Recent clinical and genetic findings. Gynecol. Endocrinol. 2009, 25, 8–11.
More