1. Actin Filaments
In HIV-1 infection, cell-to-cell viral transmission at VS has been well documented and is mainly responsible for the high decrease in T CD4+ lymphocytes during the acute phase but especially during the AIDS phase of infection, thus having clinical importance
[1]. This manner of infection involves different pathways but includes events in which an infected cell makes contact with a noninfected cell, transmitting the virus in different ways: during VS (transmission or transfer) and by cytoskeletal structures, such as filopodia or nanotubes
[1][2][3]. Hence, during viral egress, AFs stand out as scaffolds for the virus to bud. The plasma membrane is able to form structures such as filopodia or nanotubes for cell-to-cell viral transmission, as filopodia extend through receptor-mediated mechanisms from uninfected cells towards the infected cell, and a similar retroviral surfing has been described over nanotubes that also transfer viral proteins inside
[2][4][5][6][7][8][9][10][11][12]. In this case, receptor specificity seems to play a minor role, as these actin-driven structures seem to extend from HIV-1-infected cells to target cells in the absence of receptor–Env interactions
[2][4][7][8][9][10][11][13]. In DCs, by budding at the tip of filopodia, HIV-1 can tether several neighboring CD4+ T cells, leading to viral transfer and infection of the targeted T cells
[14]. Nanopores are structures formed between cells, approximately 100 nm in length, and depend on microfilament structures
[15] that, in the context of HIV-1 VS, are formed in the membrane of the target cell in which the virus “surfs” through that structure to infect and in an irrespective manner of receptor–HIV-1 Env interaction
[5]. Infection-mediated nanotubes could be established between T CD4+ lymphocytes (infected and noninfected pairs) or between T CD4+ lymphocytes and dendritic cells (DCs)
[5]. In this matter, during the infection of DCs, HIV-1 (which enters the cell using the lectin DC-SIGN) mediates the activation of a GTPase and the remodeling of the actin cytoskeleton to promote filopodia extension that allows virus transmission to neighboring CD4+ T cells
[5][14].
The ERM actin-adaptors also have relevance in HIV-1 production
[16], since these proteins connect cortical F-actin with integral and peripheral membrane proteins they are incorporated into virions, interacting with cellular components of the virological presynapse. Phosphorylation activates ezrin, which specifically accumulates at the HIV-1 presynapse in T-cell lines and primary CD4+ lymphocytes
[16]. Moreover, while cells did not tolerate a complete knockdown of ezrin, even a modest reduction in ezrin expression of approximately 50% in HIV-1-producing cells led to the release of particles with impaired infectivity. Furthermore, when cocultured with uninfected target cells, ezrin-knockdown producer cells displayed reduced accumulation of the tetraspanin CD81 at the VS and formed syncytia. Such an outcome is likely not optimal for virus dissemination, as evidenced by the fact that, in vivo, only relatively few infected cells form syncytia. Thus, ezrin likely helps secure efficient virus spread not only by enhancing virion infectivity but also by preventing excessive membrane fusion at the VS
[16]. Likewise, moesin regulates HIV-1 Env/CD4-mediated pore fusion formation in a cell-to-cell VS model, where moesin phosphorylation and its actin/plasma membrane-anchoring activity are required
[17].
Recently, the factor EWI-2, a protein that was previously shown to associate with ezrin and tetraspanins, has been identified as a host factor that contributes to the inhibition of HIV-1 Env-mediated cell–cell fusion
[18]. It has been observed that EWI-2 accumulates at the presynaptic terminal where it contributes to the fusion-preventing activities of the other viral and cellular components, and EWI-2 was also downregulated upon HIV-1 infection, most likely by the accessory viral protein U (Vpu)
[18]. In this sense, the expression levels of EWI-2 and CD81 molecules are restored on the surface of HIV-1 Env-driven syncytia
[18], where they act as fusion inhibitors, thereby providing novel insight into how deathly syncytia (i.e., collapsing by apoptosis
[19][20][21]) might be prevented from fusing indefinitely. This ‘syncytial apoptosis’ is frequently detected during HIV-1 infection in vitro and in vivo in tissues from HIV-1-infected patients
[20]. Hence, and in contrast to potential EWI-2/CD81 inhibitors
[18], it has been proposed that the induction of selective death of HIV-1-elicited syncytia might lead to the elimination of viral reservoirs. Thus, specific drugs or strategies aligned with this concept would constitute a therapeutic complement to current ARTs
[21].
One key factor that could associate cortical AFs with the cell surface and appears to regulate late stages of the HIV-1 infection cycle is the interferon (IFN)-induced type II membrane glycoprotein tetherin, also known as bone marrow stromal cell antigen 2 (BST2), HM1.24 or CD317
[22][23]. Tetherin is a GPI anchor protein localized in lipid raft microdomains that can link rafts with the underlying cortical actin cytoskeleton
[24] and acts as a direct physical tether for virions, linking the HIV-1 Env complex to the plasma membrane and preventing the final “escape” of viruses from the infected producer cell
[25][26][27][28][29][30]. Tetherin thus severely limits the spread of cell-free viruses and reduces the cell-to-cell spread of HIV-1 to a lesser degree
[31][32]. HIV-1 uses its Vpu protein to counteract the BST2/tetherin barrier
[33][34]. Vpu localizes predominantly to the trans-Golgi network (TGN) and recycling endosomes
[35][36]. Thus, Vpu antagonizes tetherin in a post-ER compartment
[37], downregulating BST2/tetherin from the cell-surface
[34] by recruiting a βTRCP2-SCF-Cullin1 ubiquitin ligase complex
[38][39][40][41][42][43].
P-selectin glycoprotein ligand 1 (PSGL-1) is another important factor recently related to HIV-1 replication that could restrict actin dynamics and arrest viral Env at the plasma membrane, resulting in virions with poor Env incorporation and reduced infectivity
[44]. In fact, PSGL-1 is a dimeric mucin-like glycoprotein that binds to P-, E-, and L-selectins and plays a role in leukocyte rolling on endothelial surfaces prior to transmigration
[45][46][47][48][49][50], being identified as an IFN-γ-induced restriction factor in CD4+ T cells using a genome-wide proteomic screen
[51]. In the context of HIV-1 infection, PSGL-1 seems to be recruited to the sites where HIV-1 particles assemble
[52]. Likewise, it seems that PSGL-1 inhibits reverse transcription in target cells and diminishes the infectivity of released virions. This inhibitory effect could be antagonized by the HIV-1 Vpu protein through the ubiquitination and proteasomal degradation of PSGL-1. Therefore, PSGL-1 could be incorporated into HIV-1 virions, exerting a negative effect on HIV-1 transmission, not only by inhibiting Env processing and incorporation but also by directly inhibiting virion attachment to target cells
[53].
A recent study concluded that HIV-1 molds AFs cortical nodes in areas of high positive membrane curvature within Arp2/3-dependent F-actin filopodia and lamellipodia structures, which enables HIV-1 to bud at the cell edge
[54]. This work is supported by live cell imaging and focused ion beam scanning electron microscopy (FIB-SEM), which permit the observation of F-actin structures that exhibit strong positive curvature where HIV-1 buds
[54]. Likewise, virion proteomics, gene silencing, and viral mutagenesis supported not only the functional involvement of Arp2/3 but also the involvement of the Rac1- and Cdc42-IQGAP1 pathways in driving F-actin regulation and membrane curvature, thus allowing HIV-1 to egress. HIV-1 also activates the Cdc42-Arp2/3 filopodial pathway for release, requiring the scaffolding protein IQGAP1 (a Rac1 and Cdc42 binding partner)
[55][56], a process that similarly occurs during cell-to-cell viral spreading
[54].
The HIV-1 structural Pr55
Gag polyprotein could be coimmunoprecipitated with actin but not tubulin, where a direct physical interaction between HIV-1 Pr55
Gag and F-actin via the nucleocapsid (NC) domain of Pr55
Gag has been reported
[57]. Likewise, it is known that there is a physical and tight association between HIV-1 Pr55
Gag and actin both in fixed and adherent cell lines
[58]. In addition, several studies have observed a high association of F-actin with HIV-1 budding sites, as some researchers have observed NC-dependent actin structures that emanate from HIV-1 buds during viral assembly and disappear upon viral release
[59]. Additionally, the presence of AFs near budding sites was later confirmed and proven to have direct physical contact with nascent viral particles
[60][61] (
Figure 1). However, some observations argue against a functional role of these microfilaments in viral budding
[61][62], but since these studies did not measure the actin content in cell-free viral supernatants, a role for actin in HIV-1 egress could not be ruled out. Therefore, given the importance of viral RNA binding to the NC region of the Pr55
Gag polyprotein during the viral life cycle, functional characterization of the NC-actin association and how this association is influenced by the viral RNA+ genome should be further addressed to understand the role played by AFs in HIV-1 Pr55
Gag packaging and virion release.
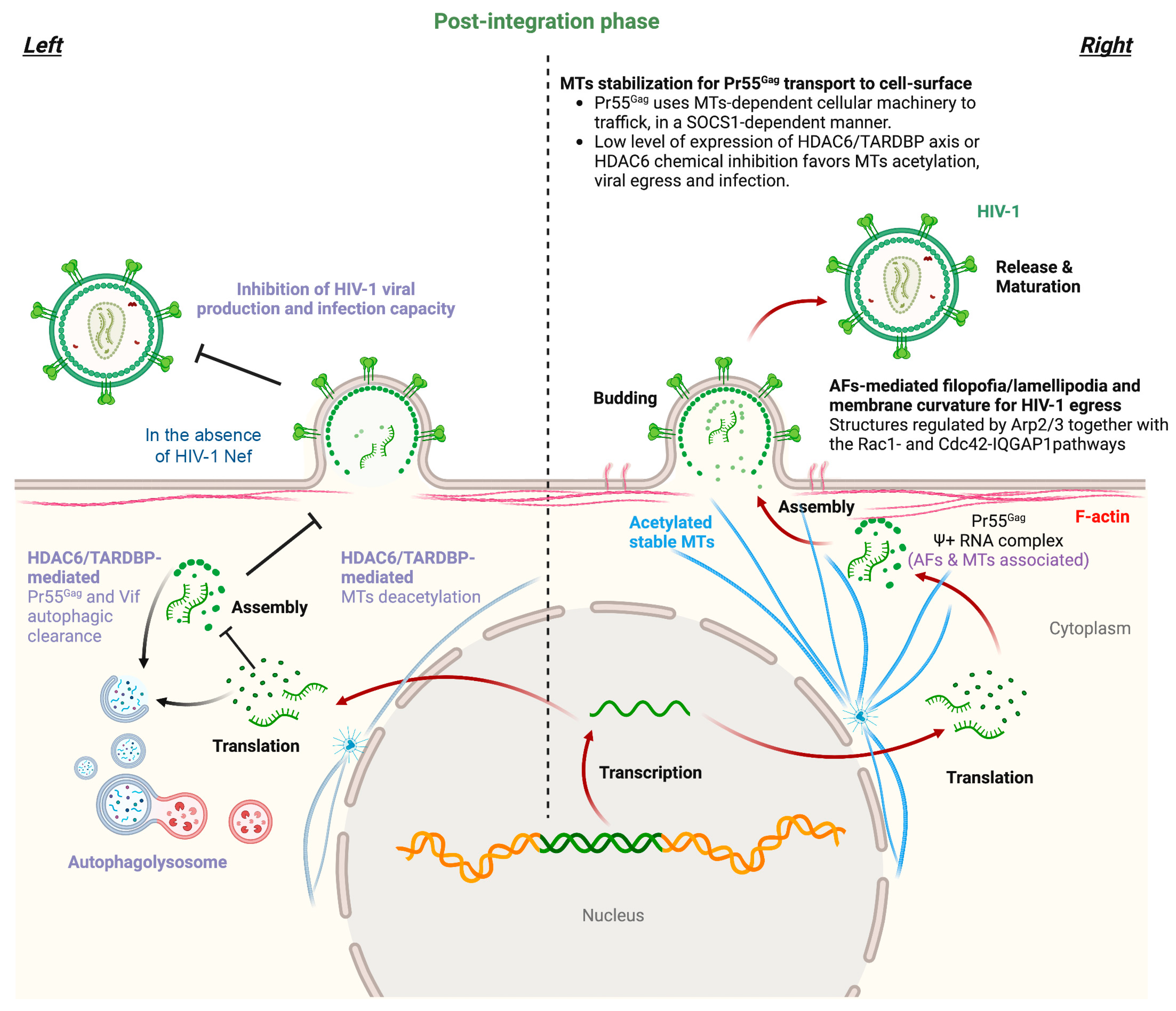
Figure 1. Scheme illustrating the late stages of the HIV-1 life cycle and associated tubulin and actin cytoskeleton dynamics. The transcription and translation of the integrated HIV-1 genome generate viral RNA+ and proteins, with the structural Pr55Gag polyprotein being relevant to recognize viral RNA+ and recruit a complex to stable MTs to travel to the plasma membrane (right panel). In fact, HIV-1 Pr55Gag uses MTs-dependent cellular machinery to traffic to the cell surface in a SOCS1-dependent manner. Pr55Gag also interacts with F-actin, thus assembling at the cell surface in areas where AFs are remodeled by the Arp2/3- and Rac1/Cdc42/IQGAP1 pathways, inducing F-actin filopodia and lamellipodia structures that finally generate high positive membrane curvatures where HIV-1 egresses (right panel). The MT-associated HDAC6 enzyme deacetylates stable MTs, thereby impeding Pr55Gag cell surface localization (left panel). Likewise, HDAC6 tubulin-deacetylase function is required to target HIV-1 Pr55Gag and Vif proteins for autophagy degradation, thus inhibiting viral production and infection capacity (left panel). HIV-1 Nef counteracts these antiviral actions by targeting the MT-associated enzyme, thereby stabilizing MTs and the HIV-1 Pr55Gag and Vif proteins. Therefore, HIV-1 Nef promotes the transport of Pr55Gag to the cell surface where it assembles to form viral particles that incorporate Vif to ensure the infectivity of HIV-1. Designs and templates were created with BioRender.
A summary of the main data discussed in this section is presented in Table 1.
Table 1.
Cellular and viral factors that influence actin cytoskeletal dynamics during the late stages of the HIV-1 viral cycle.
| Cell Factor |
Function of the Cellular Factor |
Impact on HIV-1 Infection 1 |
| ERM |
Secure efficient virus spread not only by enhancing virion infectivity but also by preventing excessive membrane fusion at the virological synapse (VS). |
+ |
| EWI-2/CD81 |
Difficult HIV-1 Env-mediates cell-to-cell fusion. |
− |
| BST2/tetherin |
Links AFs with plasma membrane where it sequesters the viral Env and preventing the virus escape from cell. |
− |
| PSGL-1 |
Inhibits RT in target cells and diminished the infectivity of released virions. |
− |
| Arp2/3 and Rac-1/Cdc42- IQGAP1 pathways |
Remodeling of cortical AFs in areas of high positive membrane curvature, within Arp2/3- and Rac1/Cdc42/IQGAP1-dependent F-actin filopodia and lamellipodia structures, which enables HIV-1 to bud and cell-to-cell spreading. |
+ |
[79][80]. Overexpression of IQGAP1 inhibits HIV-1 budding, while depletion of IQGAP1 enhances HIV-1 particle release
[77]. In fact, IQGAP1 directly interacts with both the NC and p6 regions of the structural Pr55
Gag viral protein, altering and impairing its distribution at the plasma membrane, thereby being responsible for the inhibition of viral release
[77] (
Figure 21).
An example of HIV-1 viral factors that play an important role in the late stages of the viral cycle and interact with tubulin cytoskeleton are the accessory Tat (transactivator of transcription) and viral protein R (Vpr) proteins
[81][82].
It has been observed that HIV-1 Tat interacts with monomeric tubulin and MTs. This Tat–MT association does not appear to alter or be required for Tat secretion or uptake. However, the association of Tat with MTs induces apoptosis, as the combination of Tat and MT leads to the alteration of MT dynamics and activation of a mitochondria-dependent apoptotic pathway
[81]. In addition, Bim facilitates Tat-induced apoptosis, since Bim is a proapoptotic Bcl-2 relative and a transducer of death signals initiated by perturbation of MT dynamics
[81]. Therefore, the proapoptotic Tat–MT interplay could account, in part, for the immune cell dysfunctions and pathogenesis associated with HIV-1 infection. HIV-1 Vpr is able to interact with MT-associated factors, such as EB1, p150
Glued, and dynein heavy chain
[82], and alters the MT plus end localization of EB1 and p150
Glued, negatively affecting the centripetal movement of phagosomes and their maturation
[82]. Moreover, Vpr appears to target MT/dynein-dependent endocytic trafficking in HIV-1-infected macrophages, which deeply alters phagolysosome biogenesis
[82]. It is plausible that this Vpr-provoked cell dysfunction could be toxic for infected cells by acting on caspases
[83][84][85], and mitochondria-associated factors (reviewed in
[86][87][88]) that could be activated after phagolysosome disruption
[89].
A summary of the main data discussed in this section is presented in Table 2.
It is thought that HIV-1 release and cell-to-cell transfer rely on intact and stable acetylated MTs
[63][64][65][66], such as through VS, where during transfer, the MTOC in HIV-1-infected T cells, but not in target cells, polarizes towards the VS
[65][66][67]. Notably, in infected peripheral blood lymphocytes (PBLs), the stabilization of MTs by chemical inhibition of the tubulin-deacetylase HDAC6 increases HIV-1 viral production and replication for several days postinfection
[63]. This fact indicates the importance of acetylated stable MTs for the late stages of the HIV-1 life cycle and points to the regulatory role of HDAC6 in HIV-1 replication.
In this sense, the tubulin-deacetylase HDAC6 appears to be crucial in the control of HIV-1 production and the infection capacity of nascent viral particles
[63][68][69][70]. HDAC6 directly interacts with A3G (apolipoprotein B mRNA-editing enzymatic polypeptide-like 3G or APOBEC3G) to undergo cellular codistribution along MTs and the cytoplasm in an A3G/HDAC6 complex
[70]. HDAC6 competes for Vif-mediated A3G degradation, also accounting for the steady-state A3G expression level. In fact, HDAC6 directly interacts with and promotes Vif autophagic clearance due to its C-terminal BUZ domain, a process requiring the tubulin-deacetylase activity of HDAC6
[70]. HDAC6 degrades Vif but does not affect core-binding factor β (CBF-β), a Vif-associated partner reported to be key for Vif-mediated A3G degradation. Thus, HDAC6 antagonizes the proviral activity of the Vif/CBF-β-associated complex by targeting Vif and stabilizing A3G. Finally, in cells producing virions, a good correlation was observed between the ability of HDAC6 to degrade Vif and restore A3G expression, suggesting that HDAC6 controls the amount of Vif incorporated into nascent virions and the ability of HIV-1 particles to be infectious
[70]. The tubulin-deacetylase HDAC6 also promotes the autophagic degradation of the viral polyprotein Pr55
Gag to inhibit HIV-1 production
[69]. The HIV-1 Nef factor counteracts this antiviral activity of HDAC6 by inducing its cellular clearance and subsequently stabilizing Pr55
Gag and Vif viral proteins. HIV-1 Nef interacts with HDAC6, colocalizing on MTs and neutralizing HDAC6 by degradation in an acidic/endosomal-lysosomal pathway
[69]. HIV-1 Nef therefore assures Pr55
Gag location and aggregation at the plasma membrane, promoting virus production and enhancing the infectivity of viral particles by the stabilization and uptake of Vif
[69]. Moreover, a recent study showed that the overexpression of TDP-43 in virus-producing cells stabilizes HDAC6 (at the mRNA and protein levels) and triggers the autophagic clearance of HIV-1 Pr55
Gag and Vif proteins
[68]. These events inhibit viral particle production and impair virion infectiveness, resulting in a reduction in the amount of Pr55
Gag and Vif proteins incorporated into virions.
[68]. A nuclear localization signal (NLS)-TDP-43 mutant is not able to control HIV-1 viral production and infection. Likewise, specific TDP-43 knockdown reduces HDAC6 expression (i.e., mRNA and protein) and increases the expression levels of HIV-1 Vif and Pr55
Gag proteins, thereby stabilizing MTs by α-tubulin acetylation
[68]. Thus, TDP-43 silencing favors virion production and enhances virus infectious capacity, thereby increasing the amount of Vif and Pr55
Gag proteins incorporated into virions. Notably, there is a direct relationship between the content of Vif and Pr55
Gag proteins in virions and their infection capacity
[68] (
Figure 21).
Furthermore, the concomitant PTM of MTs by acetylation produced after silencing the HDAC6/TDP-43 axis could stabilize Pr55
Gag, facilitating its transport to the plasma membrane. In this sense, it has been reported that the Pr55
Gag polyprotein colocalizes, early after expression, with viral RNA through the cis-acting packaging element, psi (Ψ), in the perinuclear region and centrioles. Ψ+ RNA and Pr55
Gag are subsequently transported to the plasma membrane of the cell
[58]. Moreover, HIV-1 Pr55
Gag uses MT-dependent cellular machinery to traffic to the cell surface in a manner dependent on the suppressor of cytokine signaling 1 (SOCS1)
[71], where stable MTs are key for SOCS1/Pr55
Gag transport
[71]. SOCS1 is a negative regulator of cytokine signaling
[72] and interacts with the MA and NC regions of Pr55
Gag through its central SH2 domain to facilitate Pr55
Gag intracellular trafficking and stability, thereby regulating the late stages of the virus replication pathway
[73] (
Figure 21).
In contrast, some MTs-associated molecules, such as the IQGAP family of scaffold proteins, which also regulate the actin cytoskeleton
[21][74][75][76], have been reported to exert an inhibitory effect on HIV-1 budding and maturation by targeting Pr55
Gag [77]. IQGAP1 has been shown to interact with the budding of enveloped viruses, as is the case for HIV-1
[78]3. Intermediate Filaments
The distribution of vimentin IFs around the nucleus and their extension throughout the cytoplasm provide a scaffold for cellular components (reviewed in
[90]) and allow vimentin to play an essential role in cellular cargo transportation
[91][92][93][94][95][96][97][98][99][100], as well as in the trafficking of viral components to the location of cell assembly and egress (reviewed in
[101][102]). It is interesting to note that vimentin IFs are distributed in cells by MTs-dependent dynamic transport
[103][104][105][106][107], regulated by AFs and related kinases
[108], which, in turn, is required to maintain the cytoskeleton network
[109][110]. Vimentin IFs modulate the replication, assembly, and egress of viruses in the host due to their known function of regulating endosomal trafficking via Rab7a and Polo-like kinase 1 (Plk1). Rab7a, which is ubiquitously present in early and late endosomes
[111][112], interacts with insoluble and soluble vimentin
[113][114]. Rab7a interacts directly with vimentin, and this interaction modulates vimentin phosphorylation and assembly
[113]. Rab7a-depleted cells have an abundance of insoluble vimentin IFs and defective endosomal trafficking
[115]. Phosphorylation of vimentin IFs at Ser
459 by Polo-like kinase 1 (Plk1) inhibits endolytic fusion during mitosis
[96]. Altogether, these interactions demonstrate that vimentin is a critical regulator of late endocytic trafficking and egress of viral particles
[116][117][118][119].
In the context of HIV-1, the knockdown of the vimentin protein has been associated with a reduction in the HIV-1 p24 capsid (CA) protein (derived from Pr55
Gag) level with concomitant impairment of viral replication
[120]. Likewise, the intracellular administration of a peptide that modifies vimentin IF distribution similarly inhibits HIV-1 replication
[120]. Moreover, the interplay of vimentin IFs with the Mac-2-binding protein (M2BP
[121]), a protein that is secreted in HIV-1-infected patients and correlates with viremia progression or control
[122][123][124], mediates the association of vimentin IFs with HIV-1 Pr55
Gag [125]. Thus, vimentin/M2BP interplay negatively affects the trafficking of HIV-1 Pr55
Gag towards the plasma membrane, whereas the absence of M2BP favors the cell surface localization of the HIV-1 Pr55
Gag polyprotein
[125][126].
It is interesting to note that some clinical studies with HIV-1 patients suggest vimentin as a potential marker for ART treatment efficiency, since in HIV-1-positive peripheral blood mononuclear cells (PBMCs) isolated from patients, vimentin was verified to be related to ART treatment and viremia control
[127]. Although there are certain limitations of this
res
tudyearch, the reported data could be of interest. For example, the protein–protein interaction analysis was database dependent, and limited clinical samples were obtained to verify the expression level of vimentin in plasma under different experimental conditions
[127]. Moreover, several reports point out that some viruses could rearrange vimentin filaments (reviewed in
[101]), and in the case of HIV-1, some published data support the possibility that vimentin may serve as a substrate for HIV-1 proteins within infected cells
[128]. In this sense, the IFs vimentin, desmin, and glial fibrillary acidic proteins are cleaved in vitro by the HIV-1 PR enzyme
[128]. Thus, microsequencing analysis showed that HIV-1 PR cleaved both human and murine vimentin. Microinjection of HIV-1 PR into cultured human fibroblasts resulted in a 9-fold increase in the percentage of cells with an altered and abnormal distribution of vimentin IFs
[128]. The ability of the HIV-1 PR to liberate amino-terminal peptides from vimentin, representing one of the best substrates of the viral PR, may be important to achieve the replication cycle because PR-derived vimentin fragments disrupt IFs organization
[129][130]. In the case of HIV-1 Vif, this accessory protein is found in the cytoplasm and in the perinuclear area colocalizing with vimentin, thereby disrupting the vimentin network
[131]. In contrast, Vif presents a diffuse pattern of distribution in the cytoplasm, nuclear membrane and nucleus when the vimentin network is chemically blocked
[131].
Most commonly, when the vimentin network is perturbed, their IFs collapse into a clump with a juxtanuclear localization. Similarly, amino-terminal polypeptides of vimentin are responsible for changes in nuclear architecture associated with HIV-1 PR activity in tissue culture cells isolated from infected individuals, and condensation or degeneration of nuclear chromatin has been described in cells from a variety of tissues
[132]. Furthermore, infection with HIV-1 includes an increase in the incidence of cancer, particularly lymphomas. Attempts at identifying an additional agent responsible for non-Hodgkin’s lymphoma in a large HIV-1-infected cohort have failed
[133], raising the possibility that HIV-1 itself is directly responsible. The ability of the HIV-1 PR to liberate amino-terminal peptides from vimentin may interfere with host cell gene expression
[128][134] and may be important to achieve the replication cycle because PR-derived vimentin fragments disrupt IF organization
[129][130]. In the case of HIV-1 Vif, this accessory protein is found in the perinuclear area colocalizing with vimentin, thereby disrupting the vimentin network
[131]. Altogether, these data indicate that the relevance of the HIV-1 Vif/vimentin IF interplay for HIV-1 replication remains to be elucidated.