Skeletal muscle is the protein reservoir of our body and an important regulator of glucose and lipid homeostasis. The dystrophin gene is the largest gene and has a key role in skeletal muscle construction and function. Mutations in the dystrophin gene cause Duchenne and Becker muscular dystrophy in humans, mice, dogs, and cats. Duchenne muscular dystrophy (DMD) is an X-linked neuromuscular condition causing progressive muscle weakness and premature death. β-hydroxy β-methylbutyrate (HMB) prevents deleterious muscle responses under pathological conditions, including tumor and chronic steroid therapy-related muscle losses. The use of HMB as a dietary supplement allows for increasing lean weight gain; has a positive immunostimulatory effect; is associated with decreased mortality; and attenuates sarcopenia in elderly animals and individuals.
1. Introduction
Over the course of three decades, the effects of β-hydroxy-β-methylbutyrate (HMB), a metabolite of leucine, on skeletal muscle mass and strength have been extensively studied
[1]. HMB forms endogenously through leucine’s reversible transamination to α-ketoisocaproic acid (KIC), catalyzed by branched-chain amino acid (BCAA) transaminase, primarily situated within skeletal muscle. The body’s production of KIC accounts for roughly 15% of the turnover rate of leucine, an essential amino acid crucial for various physiological processes. In comparison, the production of HMB, a metabolite derived from KIC, constitutes less than 1% of KIC turnover and a mere 0.1% of leucine turnover. Notably, the overall synthesis of HMB is estimated at approximately 4 μmol/kg of lean body mass per day. These metabolic proportions emphasize the relatively small but impactful role of HMB in the broader turnover of leucine and its subsequent metabolites
[2]. HMB is believed to have several mechanisms of action, including the stimulation of the mammalian target of rapamycin (mTOR), which results in increased protein synthesis
[3]. Moreover, HMB exerts an effect on protein degradation by inhibiting the ubiquitin-proteasome proteolytic pathway in muscle cells
[4]. Immobilization and catabolic conditions induce ubiquitin, leading to proteasome expression through nuclear factor kappa B (NF-κB), thereby promoting muscle wasting. NF-κB activity can lead to the expression of proteasome through ubiquitin, promoting muscle wasting. However, HMB has been shown to reduce muscle loss by inhibiting the activity of NF-κB
[5]. The efficacy of HMB supplementation has been studied in a variety of clinical conditions characterized by loss of skeletal muscle mass and weakness, including HIV (human immunodeficiency virus)
[6], critical illness
[7], and aging
[8].
Muscular dystrophies are a heterogeneous cluster of hereditary disorders that affect skeletal muscles and are primarily characterized by the progression of muscle weakness and degeneration. This, in turn, leads to a reduced lifespan of the affected individuals. The prevalence of muscular dystrophy in the general population can vary significantly depending on the specific type of muscular dystrophy and geographical region. A systematic literature review, encompassing studies classified as having a low risk of bias, reported a total combined prevalence ranging between 19.8 and 25.1 cases per 100,000 person/year for all types of muscular dystrophies
[9][12]. Among children, the most common muscular dystrophy is Duchenne muscular dystrophy (DMD), which is an X-linked inherited disease
[10][13] caused by the absence of the protein dystrophin, which is encoded by the DMD gene. DMD prevalence and birth prevalence estimates are variable throughout the literature, ranging from 0.9 to 16.8 per 100,000 males from 1.5 to 28.2 per 100,000 live male births, respectively
[11][14]. The mutation in the DMD gene disrupts the synthesis of this protein, which plays a crucial role in muscle contraction, and its deficiency results in structural and signaling malfunctions, leading to the gradual breakdown of muscular fibers and chronic inflammation, which is a significant feature of this disease
[12][15]. During the process of inflammation, neutrophils that are present in the bloodstream move toward the affected area, followed by the monocytes and lymphocytes
[13][16]. These cells release cytokines such as tumor necrosis factor-alpha (
TNF-α), interleukin 1 (
IL1), and
IL6, which are classified as proinflammatory; and
IL10, which is classified as an anti-inflammatory cytokine. The transcription factor NF-κB regulates the release of all these cytokines. This factor may be involved in the pathogenesis of DMD. Skeletal muscle inflammation is regulated by cytokine homeostasis
[14][17], However, in boys with DMD, there is an imbalance in the release of
TNF-α,
IL1, and
IL6, which are increased, and
IL10, which is decreased, as compared to healthy individuals
[15][18], indicating an imbalance in the pro- and anti-inflammatory activity. The chronic inflammation described in DMD could be associated with decreased muscle function, repetitive cycles of degeneration/regeneration, and muscle atrophy
[16][17][18][19,20,21]. Children due to their muscle weakness usually are forced to use a wheelchair by the age of 12 and die early in their third decade of life from respiratory or cardiac failure
[19][22].
The recommended medical treatment for DMD involves the administration of glucocorticoids, which act by diminishing inflammation through the reduction of inflammatory cell counts in impaired muscle fibers
[20][23]. Nevertheless, glucocorticoids carry the potential for significant deleterious side effects. Thus, alternative therapeutic approaches aimed at mitigating the inflammatory process induced by muscle fiber impairment in DMD patients are required to diminish subsequent muscle damage. Payne et al. (2006)
[21][24] studied the efficiency of HMB in a mouse model of DMD and found that HMB significantly decreases plasma creatine kinase (CK, DMD enzymatic indicator of muscle damage)
[21][24]. Since 1994, HMB holds U.S. Patent 5,348,979 “Methods of Promoting Nitrogen Retention in Humans”. There are also international patents, mostly European, that correspond to U.S. patents. The use of HMB in Poland as a dietary supplement in humans was approved by the Polish Authorities in 1996. Soon after, HMB was offered for young boys with genetically diagnosed DMD. Beneficial effects of HMB administration were observed from the very beginning of HMB treatment (unpublished). Parents of sick kids reported an improvement in their children’s motion, slower progression of a disease, better tolerance for exercise, and improved walking ability. In all HMB-treated children, there was a significant decrease in the activity of serum CK when compared with patients not treated with HMB. The oldest patient who is still successfully taking HMB (since the first symptoms of the disease) is now 32 years old. To date, more than a dozen individuals with DMD have been taking HMB supplements, with some of them having taken it for more than 20 years now (unpublished).
2. Common Pathways
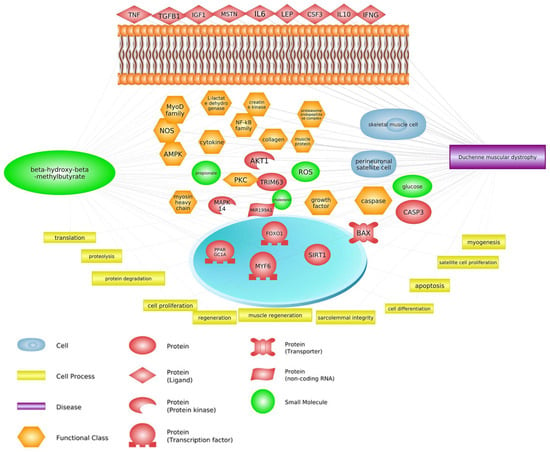
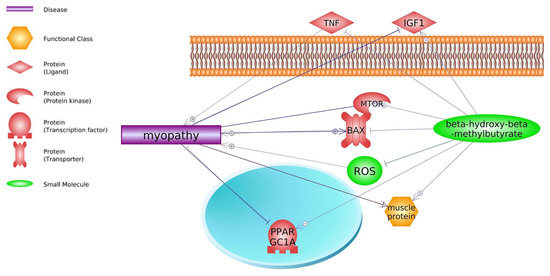
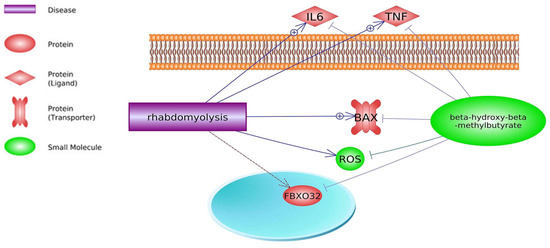
Pathway Studio Web Mammalian (V. 12.5.0.2, 2022) software analysis allowed for the identification of genes involved in both the course of DMD and the mechanism of action of HMB (keywords: Duchenne muscular dystrophy, β-hydroxy-β-methylbutyrate, and processes listed in
Figure 1,
Figure 2,
Figure 3 and
Figure 4).
Figure 1. Relevance network of genes, transcription factors, small molecules, functional classes, miRNA, and biological processes that are shared between DMD and HMB (based on Pathway Studio Web Mammalian). Tumor Necrosis Factor (TNF), Transforming Growth Factor Beta 1 (TGFB1), Insulin-Like Growth Factor 1 (IGF1), Myostatin (MSTN), AKT Serine/Threonine Kinase 1 (AKT1), Leptin (LEP), Interleukin 6 (IL6), Caspase 3 (CASP3), Mitogen-Activated Protein Kinase (MAPK14), Myogenic Differentiation (MyoD), Nitric Oxide Synthase (nNOS), Nuclear Factor Kappa B Subunit (NF-κB), Lactate Dehydrogenase (LDH), Interleukin 10 (IL10), Colony Stimulating Factor 3 (CSF3), Tripartite Motif Containing 63 (TRIM63), Interferon Gamma (IFNG), Forkhead Box O1 (FOXO1), Myogenic Factor 6 (MYF6), Caspase 3 (CASP3), BCL2 Associated X, Apoptosis Regulator (BAX), PPARG Coactivator 1 Alpha (PPARGC1A), Reactive Oxygen Species (ROS), Protein Kinase C (PKC), AMP-activated Protein Kinase (AMPK).
2.1. mTOR Signaling Pathway
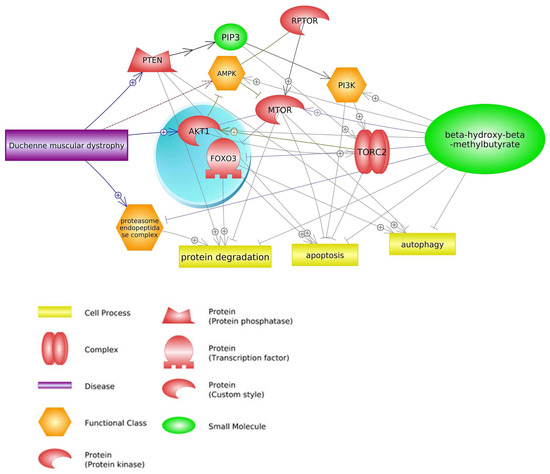
The regulation of cellular growth in response to a variety of stimuli, including energy, nutrients, and growth factors, is governed by the mTOR pathway. This pathway is primarily managed by the mammalian target of the rapamycin protein, which is classified as a member of the phosphoinositide 3-kinase-related protein kinase (PIKKs) family. mTOR assembles into two complexes with distinct inputs and downstream effects. mTOR complex 1 (
mTORC1) interacts with a novel protein named RAPTOR (regulatory associated protein of mTOR) that regulates cell growth by promoting translation, ribosome biogenesis, and autophagy. mTORC2 responds primarily to growth factors, promoting cell-cycle entry, cell survival, actin cytoskeleton polarization, and anabolic output. mTORC2 is regulated by insulin, growth factors, plasma factors, and the number of nutrients
[22][23][24][25][26,27,28,29].
One of the main factors that has a direct effect on mTOR signaling pathways is HMB supplementation, which leads to the inhibition of protein degradation, apoptosis, and autophagy
[26][27][28][30,31,32]. The findings of Baier et al. (2009)
[29][33], Stout et al. (2015)
[30][34], and Ellis et al. (2019)
[31][35] may be attributed to the anabolic properties of HMB, which activates the protein rapamycin in humans. This pathway activation leads to an increase in mRNA translation and phosphorylation of
4EBP-1 and
p70S6k, which are mTOR effectors. This, in turn, results in the synthesis of myofibrillar protein and a subsequent increase in lean mass
[32][36]. The third mechanism of HMB action that leads to an increase in mTOR activity depends on the elevated expression of insulin-like growth factor-1 (
IGF1) and growth hormone (
GH) and leads to an increase in the differentiation and proliferation of satellite muscle cells
[33][34][11,37]. In addition, HMB can stimulate mTOR activity by the impact on
AKT serine/threonine kinase 1 (
AKT1) expression. Overexpression of
AKT1 by HMB supplementation leads to an increase in mTORC1
[35][36][38,39] and mTORC2 activities
[37][38][39][40,41,42], as well as concomitant inhibition of
AMPK expression (one of the DMD biomarkers)
[40][41][43,44]. Moreover, HMB in this pathway inhibits proteasome endopeptidase complex activity that leads to attenuation of protein degradation during DMD
[42][43][45,46] (
Figure 2). Activation of mTOR complexes leads to inhibiting DMD side effects such as apoptosis
[25][29] and protein degradation
[24][28].
Figure 2. Genes common for DMD and HMB—mTOR signaling pathways (based on Pathway Studio Web Mammalian
[44][45][46][47][48][49][50][51][47,48,49,50,51,52,53,54]). Phosphatase and Tensin Homolog (PTEN), Regulatory Associated Protein of mTOR Complex 1 (RPTOR), CREB Regulated Transcription Coactivator 2 (TORC2), AKT Serine/Threonine Kinase 1 (AKT1), Forkhead Box O3 (FOXO3), Phosphatidylinositol (3)-trisphosphate (PIP3), AMP-activated Protein Kinase (AMPK), Phosphatidylinositol 3-kinase (PI3K), Mechanistic Target of Rapamycin (mTOR). The “+” symbol at the end of the arrow corresponds to stimulation, the “┤” symbol corresponds to inhibition.
2.2. FOXO1 Signaling Pathway
The FOXO family of proteins, specifically Foxo1, are nuclear transcription factors that have a significant impact on the regulation of overall energy metabolism in the body. Their presence is ubiquitous across tissues, and they are involved in various processes that control cell proliferation and differentiation. To maintain glucose homeostasis, the production and uptake of glucose in peripheral tissues must be adjusted secretion to insulin. During periods of fasting, the liver is primarily responsible for maintaining glucose levels, and Foxo1 is instrumental in promoting the expression of gluconeogenic enzymes. FOXO1 indirectly regulates the formation of adipose tissue and skeletal muscles, particularly in the presence of metabolic dysfunction or insulin resistance. These proteins hold potential as molecules that bridge the gap between longevity and tumor suppression
[52][55].
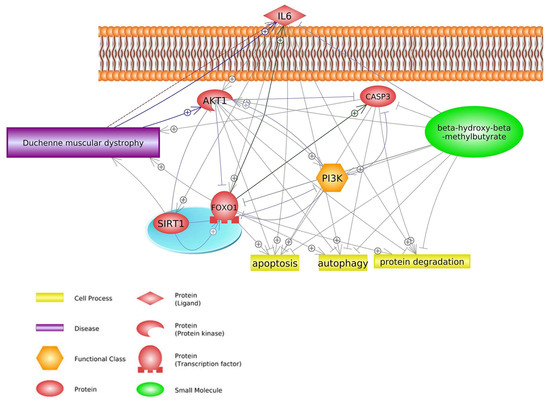
During muscle inflammation that accompanies DMD, an increase in NF-κB signaling stimulates leukocytes to release
TNF-α and proinflammatory cytokines
IL1 and
IL6 [53][57].
NF-κB plays a central role in inflammatory reactions and is the primary activator of cytokines. In contrast, HMB suppresses upregulated
IL6 production in response to
TNF-α stimulation in cancer cell lines in vitro through control of the ubiquitin-proteasome pathway without inducing apoptosis
[54][58]. HMB can inhibit some genes that lead to the progression of DMD or upregulation of this disease, for example,
IL6, a biomarker of DMD
[14][17], is overexpressed in DMD patients compared to healthy individuals
[14][55][56][17,59,60]. In contrast, HMB suppresses the expression of
IL6 in DMD disease, and consequently, apoptosis of muscular cells and DMD side effects will decline
[54][58]. It should be noted that
IL6 in the Foxo1 signaling pathway enhances Foxo1
[57][61] and Sirtuin 1 (
SIRT1)
[58][62] expression, and these factors have positive effects on DMD. Another protein that is inhibited by HMB is caspase 3 (
CASP3)
[59][10]. This protein has not only a positive effect on DMD
[60][63] but also enhances some biological processes such as apoptosis and protein degradation
[60][61][62][63,64,65] (
Figure 3).
Figure 3. Genes common for DMD and HMB—FOXO1 signaling pathways (based on Pathway Studio Web Mammalian
[14][54][60][63][64][65][66][17,58,63,66,67,68,69]). AKT Serine/Threonine Kinase 1 (AKT1), Interleukin 6 (IL6), Caspase 3 (CASP3), Forkhead Box O1 (FOXO1), Phosphatidylinositol 3-kinase (PI3K), Sirtuin 1 (SIRT1). The “+” symbol at the end of the arrow corresponds to stimulation, the “┤” symbol corresponds to inhibition.
2.3. Insulin Signaling Pathway
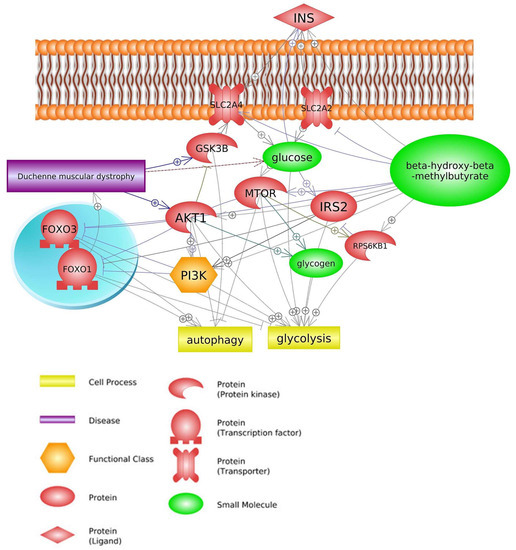
Insulin is a highly effective anabolic factor that induces cellular growth and differentiation, while also facilitating the storage of substrates in the liver, muscle, and fat. This occurs through insulin’s stimulation of lipogenesis, glycogen and protein synthesis, and inhibition of lipolysis, glycogenolysis, and protein breakdown
[67][68][69][70,71,72]. Insulin also supports glucose transport in muscle and fat cells by promoting the translocation of glucose transporter GLUT4 from intracellular sites to the cell surface. Of the insulin-dependent glucose disposal, roughly 75% occurs in skeletal muscle, while adipose tissue accounts for a relatively small fraction. Additionally, insulin activates mTOR, a member of the PI3K protein family that primarily functions as a serine kinase. The activation of this protein kinase involves PI3K activation, although another signal may also be necessary. The mammalian translation machinery can be regulated by mTOR through direct phosphorylation that activates p70 ribosomal S6 kinase and the initiation factor 4E for the eukaryotic translation inhibitor, PHAS1 or 4E-binding protein 1. As a result of p70’s activation of ribosome biosynthesis by phosphorylating the ribosomal S6 protein, mRNA translation is increased
[70][73]. The release of insulin from the pancreas leads to glucose uptake in peripheral tissues, except the liver, and switches the body from catabolism to anabolism. Insulin inhibits glycogenolysis and gluconeogenesis in the liver while stimulating glycogen accumulation through a coordinated increase in glucose transport and glycogen synthesis
[71][74].
The processes of glycolysis and glycogenolysis are suppressed in DMD patients, leading to a significant decrease in glucose levels, the main energy substrate. This energy deficit is likely the cause of the observed muscular weakness in these individuals
[72][75]. The absence of dystrophin protein in DMD patients results in abnormal glucose transport mechanisms via multicomponent glycoprotein complexes, which may contribute to the reduction in glucose concentration in dystrophic muscle.
The control of muscle protein synthesis in terms of nutrition is typically influenced by the insulin-dependent mechanism, where the release of insulin triggered by the intake of carbohydrates and/or proteins results in a nitrogen-conserving effect that facilitates a favorable net balance. However, it has been demonstrated that HMB supplementation does not elicit any impact on insulin supplementation
[73][78]. Therefore, suppression of protein breakdown could be considered insulin-independent. Indeed, HMB has been shown to suppress ubiquitin-proteasomal regulated muscle protein breakdown and inhibition of myonuclear apoptosis by antagonizing mitochondrial-associated caspases
[64][67]. Despite the inhibitory HMB effect on muscle protein breakdown, there were no obvious effects on the proteolytic markers such as
MURF1 and
MAFBX, and therefore, how and through which proteolytic pathway HMB is regulating reductions in protein breakdown remains not clear
[3][28][3,32]. Moreover, HMB has been shown to attenuate the increased phosphorylation of RNA-dependent protein kinase, which may activate
NF-κB, whereas increased phosphorylation of mTOR could lead to phosphorylation and subsequent nuclear exclusion of Foxo3
[51][54]. HMB has been demonstrated to effectively prevent the nuclear accumulation of NF-κB and enhance the phosphorylation of mTOR in reaction to catabolic stimuli, as evidenced by
Figure 4 [59][10].
Figure 4. Genes common for DMD and HMB—insulin signaling pathway (based on Pathway Studio Web Mammalian
[3][45][46][49][51][63][74][75][3,48,49,52,54,66,79,80]). Insulin (INS), Solute Carrier Family 2 Member 4 (SLC2A4), Solute Carrier Family 2 Member 2 (SLC2A2), Glycogen Synthase Kinase 3 Beta (GSK3B), Ribosomal Protein S6 Kinase B1(RPS6KB1), Insulin Receptor Substrate 2 (IRS2), AKT Serine/Threonine Kinase 1 (AKT1), Forkhead Box O1 (FOXO1), Forkhead Box O3 (FOXO3), Mechanistic Target of Rapamycin (mTOR), Phosphatidylinositol 3-kinase (PI3K). The “+” symbol at the end of the arrow corresponds to stimulation, the “┤” symbol corresponds to inhibition.
3. Common Genes
Data analysis carried out with the use of the Pathway Studio Mammal Plus software (V. 12.5.0.2, 2022), along with the analysis of literature data, allowed for the identification of aforementioned common pathways and additionally some genes of significant importance for the course of DMD disease and indicated as being under the influence of HMB (
Figure 1). The main common genes are listed in (
Table 1), along with their effect on DMD and HMB, and the literature reference. Those not mentioned above as taking part in common DMD/HMB pathways are discussed below.
Table 1. The main genes common for DMD and HMB.
| Gene |
DMD Effect |
References |
HMB Effect |
References |
| MSTN |
inhibitor |
Chiu, W. et al. (2020) [76][81] |
stimulator |
Shirvani, H. et al. (2020) [77][82] |
| AKT1 |
stimulator |
Peter, A.K.; Crosbie, R.H. (2006) [44][47],
Kornegay, J.N. et al. (2012) [78][83] |
stimulator |
Salto, R. et al. (2015) [46][49]
Schnuck, J.K. et al. (2016) [74][79] |
| LEP |
stimulator |
Söderpalm, A.C. et al. (2007) [79][84] |
stimulator |
Świetlicka, I. et al. (2021) [80][85] |
| IL6 |
stimulator |
Rodríguez-Cruz, M. et al. (2018) [14][17]
Gallardo, F.S. et al. (2021) [56][60] |
inhibitor |
Miyake, S. et al. (2019) [54][58] |
| CASP3 |
positive effect |
Parrotta, E.I. et al. (2020) [60][63] |
inhibitor |
Eley, H.L. et al. (2008) [59][10]
Hao, Y. et al. (2011) [64][67] |
| MAPK (MAPK14) |
biomarker |
Cotán, D. et al. (2016) [81][86] |
inhibitor |
Eley, H.L. et al. (2008) [59][10] |
| MyoD family |
unknown |
Gonçalves, M.A.F.V. et al. (2008 and 2011) [82][83][87,88] |
stimulator |
Kim, J.-S. et al. (2013) [43][46] |
| nNOS |
unknown |
Tian, L.J. et al. (2014) [84][89] |
stimulator |
Peterson, A.L. et al. (1999) [85][90] |
| NF-κB |
stimulator |
Wattin, M. et al. (2018) [50][53]
Finkel, R.S. et al. (2021) [86][91] |
inhibitor |
Smith, H.J. et al. (2005) [4]
Miyake, S. et al. (2019) [54][58] |
| LDH |
stimulator |
Luce, L.N. et al. (2016) [87][92]
Sadek, A.A. et al. (2017) [88][93] |
unknown |
Tsuchiya, Y. et al. (2019) [89][94] |