Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Laura Veschetti and Version 2 by Lindsay Dong.
Genomic instability comprises not only the accumulation of mutations but also telomeric shortening, epigenetic alterations and other mechanisms that could contribute to genomic information conservation or corruption.
- genomic instability
- DNA repair
- human complex disorder
- evolutionary genetics
- ncRNA
1. Introduction
A struggle of the collective drive toward complexity, auto-organization and genomic diversification against the “self” need to protect one’s own genomic information has been taking place since the beginning of life. On one plate of the scale, there is the dynamism of mechanisms involving genomic variability: gene transfer, duplication, rearrangements, recombination and exchange of mobile genetic elements, which are the events that most likely lead the drive to biological complexification [1][2][1,2]. On the other plate, there is the reliability of DNA conservation: a plethora of repair systems adapted and evolved together with organisms’ genomes to preserve them from corruption during individuals’ and cells’ lifespans and reproduction [3].
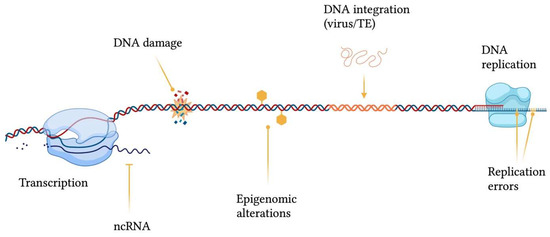
2. The Balance between Variability and Conservation
Genomic structural integrity and functional stability are constantly threatened by DNA and chromatin damaging agents ranging from exogenous sources, such as environmental toxins (e.g., polycyclic aromatic hydrocarbons), ultraviolet light (UV), ionizing radiation and mutagenic chemicals, to endogenous processes, including DNA replication and repair errors, epigenetic dysregulation, telomere shortening, spontaneous decay of DNA, transposable elements (TEs) insertions and oxidative stress (e.g., generation of reactive oxygen species) [4] (Figure 1). The consequences of damaging events are displayed as a wide variety of genomic wounds which comprehend base mismatches, single-strand breaks (SSB), double-strand breaks (DSB), inter-strand crosslinks, intra-strand crosslinks, bulky adducts and genomic rearrangements [5].Figure 1. Sources contributing to genomic instability. ncRNA = non-coding RNA; TE = transposable element. This figure was generated using BioRender.
In order to remediate DNA damage, a plethora of DNA repair mechanisms (Figure 2) have emerged across the tree of life, and their importance is evidenced by the presence of redundant, complementary and conserved repair systems. Indeed, such systems are so important that in the debated research for a minimal genome, researchers found that up to 5% of the required genes have to be committed to DNA repair mechanisms [6][7][9,10]. The rationale behind having such a wealth of repair systems lies in the fact that each mechanism is able to recognize and fix specific damage substrates [8][11]. For example, base-excision repair (BER) can resolve base mismatches, single-strand breaks, and intra-strand crosslinks by generating an apurinic/apyrimidinic site (AP site), which is then cleaved by an AP endonuclease, thus creating a single-strand break that is then closed by nucleotide synthesis [9][12]. In addition, base mismatches can also be detected and corrected by mismatch repair (MMR) together with replication slippages [10][13], whereas single-strand breaks are also repaired by SSB repair (SSBR) [11][14]. Conversely, DSB—the most deleterious form of DNA damage—can be either resolved by homologous recombination (HR), which uses a homologous DNA template for repair [12][15], or by nonhomologous end-joining (NHEJ) through DNA ends ligation [13][16]. Finally, inter-strand crosslinks can be processed via the Fanconi anaemia (FA) pathway and nucleotide excision repair (NER), which removes bulky, helix-distorting lesions, including intra-strand crosslinks [14][17].
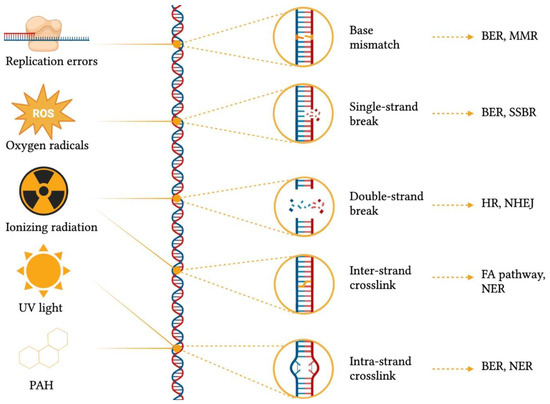
Figure 2. Common DNA damaging agents, types of genomic scars caused by different damage sources, and damage repair mechanisms. BER = base-excision repair; FA = Fanconi anaemia; HR = homologous recombination; MMR = mismatch repair; NER = nucleotide excision repair; NHEJ = nonhomologous end-joining; PAH = polycyclic aromatic hydrocarbons, ROS = reactive oxygen species; SSBR = single-strand break repair; UV = ultra-violet. This figure was generated using BioRender.
Both DNA damage and repair systems leave genomic scars typical of the mechanisms involved, thus generating genomic diversity. Such scars can either be driving forces to physiological processes—as adaptive immune response and meiosis (e.g., DSB-mediated recombination) [15][16][18,19]—or side effects of a defective restoration of information. For example, MMR deficiency may induce single nucleotide substitution and variation in the length of short repetitive DNA sequences (e.g., microsatellites) [17][20], whereas HR defects typically lead to loss-of-heterozygosity, allelic imbalances extending to the telomeres and large-scale rearrangements [18][21].
DNA damage and repair, change and conservation of information, and adaptation and selection are factors whose weights reside on opposite plates of the scale, and the balance between them is ruled by laws that still need to be fully understood. In this context, the environment emerges as a not-so-hidden judge favouring one plate over the other. A notable example is the onset of “hypermutator” phenotypes in response to environmental factors [19][20][22,23]. This phenomenon implies increased mutation rates and can be observed in several events, such as microbial adaptation and cancer evolution [21][22][23][24,25,26]. Many theories have been proposed to explain genomic instability in terms of fitness in certain environments. Focusing on microbial adaptation, the prevailing hypothesis states that genomic instability is a profitable event because it increases the microbial population’s overall chance of survival [24][25][26][27,28,29]. Breivik and colleagues proposed a shift of perspective from colony to single-cell level by focusing on the biological cost of DNA repair: genomic instability arises because DNA repair may cost more than the errors it prevents in mutagenic environments [27][30]. Even though the debate on the possible advantages brought by genomic instability is still open, unstable genomes seem to be transiently favoured in stressful environments, whereas stable ones adapt more successfully in the long run [28][29][31,32].
3. The Guardians of Genomic Stability across the Tree of Life
One of the first attempts to study the evolution of repair systems was carried out by Aravind, Walker and Koonin in 1999 [8][11]. The scientists searched for homologues of the repair proteins sequences of model organisms Escherichia coli and Saccharomyces cerevisiae in many bacteria, archaea and eukaryotes genomes. The authors reported a considerable heterogeneity in repair systems across the tree of life: they found that proteins involved in DNA repair seem to follow the “domain Lego” principle, according to which proteins are generated by copying, shuffling and recombining a limited number of conserved domains. Moreover, horizontal gene transfer—between bacteria and archaea, as well as between organellar and eukaryotic genomes—was proposed as a key mechanism contributing to the richness of repair systems.
The last universal common ancestor is supposed to have evolved in a high temperature and anoxic environment when the Earth magnetic field was still weak and the planet vulnerable to ionizing radiation. Since that time, the environment and conditions in which life develops and thrives have dramatically transformed and the revolutionary change has been the increase in oxygenation levels, determined by the evolving organisms themselves [30][33]. Indeed, before the Great Oxidation Event (around 2.4–2.0 giga-annum ago), the predominant threats to genomic integrity were base loss, cytosine deamination, and damages induced by UV light, ionizing radiation, and alkylation, whereas in today’s world oxidative DNA damage is a substantially bigger concern [31][34].
3.1. Nucleotide Excision Repair Pathway
Modern NER evolved only after the separation of bacteria and eukaryote domains and introduced the ability to repair a wide range of bulky helix-distorting DNA adducts by damage recognition, lesion excision and DNA synthesis [32][37]. In particular, NER is present in bacteria in the form of the widely conserved UvrABC protein complex: UvrA is involved in damage recognition, UvrB is a helicase that opens the dsDNA and UvrC a nuclease that operates cuts on both sides of the damage [33][38]. In eukaryotes, an analogous and more complex NER pathway can be found: XPC-hr23b performs damage recognition, transcription factor IIH opens the dsDNA and binds to XPA and RPA proteins, and XPF-ERCC1 and XPG nucleases are finally recruited to perform lesion excision [33][38].3.2. Mismatch Repair System
Another universal repair pathway is MMR which has as key players MutS and MutL proteins in bacterial species (except for Actinobacteria) and their respective homologues in eukaryotes [34][42]. Interestingly, most archaea lack MutS and MutL genes homologues and the few groups that harbour them tend to be temperature mesophiles such as halophiles and methanogens, which most likely acquired these genes via the horizontal transfer. In the great majority of archaeal organisms an alternative pathway, named endonuclease mismatch specific (EndoMS), that detects and corrects mismatches can be found [35][43]. Curiously, EndoMS is also present in bacterial genomes belonging to the Actinobacteria phylum, where MutS and MutL are absent [36][44]. MMR deficiency in humans is associated with microsatellite instability (MSI) across different cancer types as colorectal and endometrial carcinomas [37][45].3.3. Double-Strand Break Repair
Both HR and NHEJ systems are involved in the repair of DSBs. HR is one of the universal DNA repair systems and is implicated in the restart of DNA replication at stalled forks [38][54]. It is also involved in promoting genetic diversity via DNA transfer [39][40][55,56]. However, this is an energetically demanding and complex process, and, for this reason, simpler but less accurate pathways, such as NHEJ, operate alongside HR. In eukaryotic cells, NHEJ is commonly used in the G1 phase of the cell cycle since it does not depend on the presence of a homologous DNA duplex [33][38]. The NHEJ pathway has also been reported in bacteria by Weller and colleagues, who underlined the need of a DNA end-binding bacterial Ku protein for the correct operation of this system [41][57].3.4. DNA Repair: Going Viral
At the end of this excursus through the branches of the tree of life, it is necessary to include a reflection on viruses. Indeed, DNA damage response can be activated by incoming viral DNA, during the integration of retroviruses, in response to aberrant DNA structures generated upon active viral DNA synthesis, or during persistence of extrachromosomal viral genomes [42][43][60,61].4. Genomic (in)Stability in Homo sapiens: Just a Matter of Luck?
4.1. Nuclear and Mitochondrial DNA
Nuclear DNA and mitochondrial (mtDNA) are two separated genomes, indeed they present structurally different DNA molecules: the diploid linear nuclear genome and the multi-copy haploid circular mitochondrial genome. In recent decades, several studies pointed out their interconnection relatively to the handling of DNA damage [44][45][46][71,72,73]. For example, Baulch reported how genomic instability induced by radiation may alter cellular epigenetic mechanisms and can reduce mitochondrial functions; at the same time, mitochondrial dysfunction hampers the cell epigenetic profiles [47][74]. Several DNA repair mechanisms emerged throughout evolution, and more than 125 genes involved in such systems have been identified in humans [48][49][78,79]. It has been observed that the mutation rate is higher in mitochondria than in the nuclear DNA [50][51][80,81]. Specifically, mtDNA is more prone to oxidative damage due to the presence of a higher concentration of ROS—which account for approximately 10,000 daily DNA lesions per cell [52][66]—and the lack of chromatin protection [53][82]. The accumulation of mtDNA damage can lead to mitochondrial dysfunction and has been linked to age-related diseases, such as Parkinson’s disease [54][83] or Werner syndrome [55][84], and to different types of cancer [56][85].4.2. Evolutionary Insights
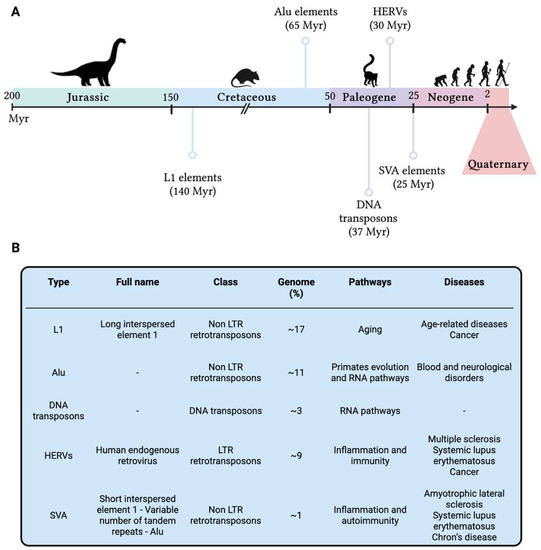
Genomic instability is gradually acquiring a central role in our knowledge on human evolution, providing novel insights on our past as humankind, as well as new perspectives on future therapeutical targets. Breivik and Gaudernack [27][30] were probably the first to hypothesize the thin trade-off intrinsic to genomic instability: the loss of genomic stability might give to evolutionary mechanisms the opportunity to take action and to explore possibilities for fitness advancement and novel adaptation [57][99], but, at the same time, it might be indirectly associated with an increased risk of several late-onset diseases [58][100], which from an evolutionary perspective are unfavourable explorations. From a similar perspective, Little gave insight on the concept of randomness, profoundly inborn in evolution and, hence, in the evolutionary trade-off concept [59][101]: not only does evolution take place in random ways but also DNA damage and genomic instability happen randomly either in time (in terms of lifetime as well as triggering causes) or in space (in terms of genomic location or cellular localisation). When it comes to humans, this novel perspective amplifies the research focus not only on present-day biological processes but also on our genomic history as human beings: all our genetic changes are written in our DNA, hence investigating DNA lesions scarred in our genomes might provide novel insights on host-pathogen co-evolution, genomic instability, and disease onset and/or severity. To better understand genomic (in)stability across the human evolution, Cordaux and Batzer [60][105] focused on the analysis of transposable elements (TEs) as major players still influencing the human genome functionality. TEs are DNA sequences that can move within the genome, originally identified in maize [61][106] and subsequently confirmed in humans [62][107]. Nearly half of the human genome seems to be composed of TEs, and this could be an underestimation due to the presence of extremely ancient TEs which are no longer recognisable [63][108]. In human, several TEs have been identified such as DNA transposons, long terminal repeats (LTRs) retrotransposons as the human endogenous retroviruses (HERVs), non-LTR retrotransposons as long interspersed element 1 (LINE-1 or L1), Alu and SINE-R-VNTR-Alu (SVA—composed of short interspersed nuclear element of retroviral origin [SINE-R], variable number of tandem repeats [VNTR], and Alu), and other minor elements [64][109]. All these elements are densely distributed along the genome and have a strong impact in shaping human genomic structural and functional features, carrying information of past adaptation as well as the seeds of evolution, either in terms of fitness improvement and genomic innovation [65][110], or as causes of genomic instability and genetic disorders [66][111]. However, TEs are currently not mobile in the human genome and their last activity is definitely far from present days. TEs were active at different ages along the evolutionary history of mammalian organisms, and their origin is dated several millions of years (Myr) ago (Figure 35).Figure 35. (A) Timeline of the activities of the primary transposable elements which had a fundamental role in the evolution of present-day humans. (B) Main transposable elements in the human genome reported in chronological order of activity. Genome = the percentage of human genome identified as TE type; Pathways = pathways in which TEs have been shown to play a role; Diseases = diseases in which TEs have been reported to be implicated. HERVs = Human Endogenous Retroviruses; L1 = Long Interspersed Nuclear Elements 1; LTR = Long Terminal Repeats; Myr = millions of years; SVA = SINE-R-VNTR-Alu. This figure was generated using BioRender.
SVA elements [67][130] are not so present in the human genome, accounting for roughly 3000 copies [68][131], probably due to their nonautonomous nature and their LINE-1-related origin [69][132]; however, like most of the previous reported TEs, SVA seems to be associated with inflammatory conditions and autoimmune diseases, such as amyotrophic lateral sclerosis [70][133], systemic lupus erythematosus and Chron’s disease [71][134]. TEs showed to be fundamental for hominoid and human evolution, shaping their development and genomic advancement for several hundreds of millions of years and resulting in an increase in size of the human genome and a significant inter-individual variability in TEs content [72][135], turning out to be a highly informative vault for human evolutionary history. Moreover, their presence in present-day humans is a strong signal of evolutionary advantage, since they have been maintained in our genome for several millennia: some examples are TEs contribution to genetic innovation, such as the introduction of new genes in the whole known lifespan of humankind [60][105] and their implication in some regulatory networks [73][136], including immunity [74][137] and embryonic development [75][138].
5. Genomic Instability, Aging and Late Onset Complex Diseases
5.1. Telomeric Instability
Human telomeres are composed of a long stretch (up to tens of kilo-base pairs) of TTAGGG nucleotide repeats located at the end of each chromosome to protect them from degradation and ensure their stability [76][77][156,157]. Indeed, cells carry a variety of mechanisms and proteins—including the shelterin complex and telomerase—responsible for the maintenance of telomeres length [77][157]. However, the mitotic process determines a shortening of telomeres in daughter cells compared to the parent cell, thus telomeres have been proposed as “molecular clocks” for aging [78][158]. Moreover, shortened telomeres trigger replicative senescence and impair the regenerative capacity of tissues, which is undesirable in the case of pluripotent stem cells and adult stem cell compartments [79][159]. Impairment of telomeric maintenance and accelerated telomere shortening have been found to be associated with some of the leading causes of disease and death; among them: central obesity [80][81][160,161], lifetime accumulation of stress [82][83][162,163], increased risk of cardiovascular events [84][85][164,165], and reduced immune response to influenza vaccination [86][166]. In particular, somatic mutations in genes involved in telomeres maintenance have been linked to the functional decline of B lymphocytes, skeletal muscle cells, and neurons [78][158].5.2. Microsatellites Structural Maintenance
Microsatellites are short tandemly repeated sequence motifs consisting of 1–6 bp that are typically repeated up to 50 times in millions of locations across the genome [87][169]. At least two main mechanisms can play a role in the failure of microsatellite structure maintenance: (1) DNA replication errors (e.g., due to polymerase slippage) that impact the length of microsatellites, and (2) defects in DNA repair mechanisms that determine an accumulation of errors leading to the generation of shorter/longer novel fragments (also known as MSI). In the first case, the number of repeat units changes from one generation to the next due to replication slippage. In particular, alleles with a higher repeat number appear to be less stable than those with a lower number of repeats, which explains why a highly significant excess (compared to the expectation under the assumption of random effect) of long microsatellites has been observed in humans and across different species [88][170]. This type of instability can affect different genomic locations with a varying magnitude, which is reflected both in repeat expansion disorder onset timeframe (i.e., the greater the damage, the earlier the onset age, also known as anticipation) and phenotypic severity (i.e., ranging from mild to severe phenotypes).5.3. Mitochondrial Dysfunction
Mitochondria are additional key players involved in aging and CD development. Actually, the role of mitochondria in aging is so determining that a “mitochondrial theory of aging” has been proposed [89][178]: with age, mitochondria accumulate ROS-induced damage and become dysfunctional, and the function of cells declines causing aging. It is apparent that mitochondrial dysfunction particularly affects organs that require high levels of energy such as the heart, skeletal muscles and brain [90][179].5.4. Human Networking: The Systemic Complexity of Life
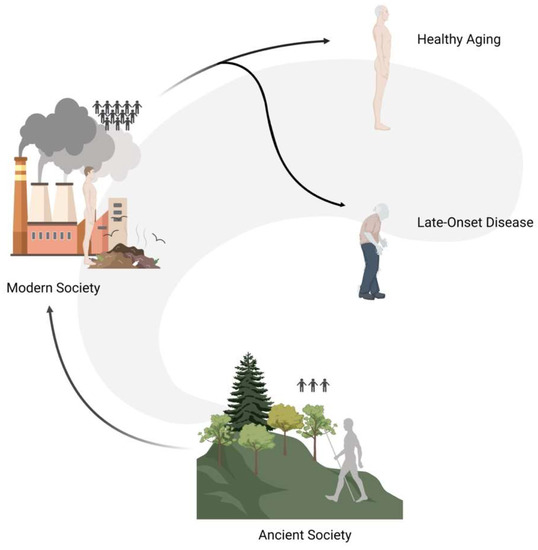
As humans, we do not live as single entities but we are a part of complex systems and communities (Figure 46). In the past, the great majority of people lived in isolated groups composed of a small number of individuals (i.e., 50–100 people), and only recently—for evolutionary times—have people started gathering in larger cities. Such transition went hand in hand with a radical change in social dynamics that had repercussions on the population genetic scale: in the past, the genetic variation pool of individual communities was very limited and selective pressures (e.g., disease, famine, etc.) were particularly high, whereas today humans make up a very extensive community with an overall rich pool of genetic variation.Figure 46. Graphical representation of social dynamics changes that had repercussions on the populations genetic level. In the past, people lived in isolated groups composed of a small number of individuals (ancient society), and only recently have people started gathering in larger cities (modern society). The modern ability to prevent the impact of selective pressures determined the maintenance of variants that in a natural setup would have been filtered out, thus possibly causing late-onset diseases.
Up to the XX century infectious diseases were the main cause of mortality, whereas, in recent decades, this role has been taken up by diseases such as cancers, cardiovascular diseases, and metabolic disorders [91][187]. This might indicate that the attenuation of selective pressures acting on human beings might allow variants with mild effects to play a detectable role in the long term, either through damage accumulation (e.g., threshold effect) or simply because they have the chance to manifest their effects with the progression of aging.