Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sihua Hou and Version 2 by Camila Xu.
Deconstructive synthesis, namely building new, challenging structures through bond cleavage of easily accessible moieties, has emerged as a useful design principle in synthesizing bioactive natural products. Divergent synthesis, namely synthesizing many natural products from a common intermediate, can improve the efficiency of chemical synthesis and generate libraries of molecules with unprecedented structural diversity.
- bioactive natural products
- total synthesis
- deconstructive synthesis
- divergent synthesis
1. Introduction
Since the synthesis of urea in 1828, humans have opened the door to organic synthesis. With the development of isolation and identification techniques and organic synthesis methods, people are pushing the limits of complex molecular synthesis, especially complex natural products, such as vitamin B12 and anemone toxin. Natural products are the main source of innovative medicines, such as artemisinin and paclitaxel. From 1981 to 2020, over 50% of the small molecule drugs approved by the FDA were derived from natural products or the parent molecular structure of natural products [1]. It can be seen that innovative drug development based on active natural products has played a crucial role in the field of original drug research and development. However, active natural products are often very scarce in nature and have low direct druggability. Therefore, research on the total synthesis of active natural products can not only solve the problem of limited natural resources and prepare a large amount of active natural products but also enable the rapid synthesis of natural product analogues with diverse skeletons and functional groups, building a natural product-like compound library to conduct extensive biological activity screening and drug development research. It has important scientific significance and potential application value in the field of natural product synthesis and new drug development. Thus, the development of practical synthetic methods and efficient synthetic strategies in natural product total synthesis has always been an active field in organic synthesis and has attracted great attention from synthetic chemists.
Deconstructive synthesis, namely building new (and often more challenging) structures through bond cleavage and the formation of easily accessible moieties, has emerged as a useful design principle in preparing complex bioactive natural products and other target molecules [2]. The basic logic of deconstructive synthesis is to construct a “hardly accessible” skeleton from an “easily accessible” skeleton through skeletal reorganization. Meanwhile, the rapid construction of polycyclic molecular frameworks, the precise instillation of multi-stereocenters bearing crowed quaternary carbon centers, and the efficient induction of versatile functional groups should also be highlighted. It also facilitates the continuous modification of key advanced intermediates to improve the overall efficiency of the synthetic route. The subtlety of deconstructive synthesis often lies in its creative approach of building intricate molecular frameworks, resulting in the design of easily accessible intermediates that could significantly reduce the challenge of total synthesis. The development of deconstructive synthesis strategies is often inspired by the molecular structural characteristics of natural products, which also tests the creativity and personalized perspective of synthetic chemists.
Divergent synthesis, in short, involves working from one common intermediate to many natural products (Figure 1). In general, a class of natural products often shares a similar chemical structure with various functional groups or different molecular frameworks that can be constructed from a common intermediate in biosynthesis. The original definition of divergent synthesis was proposed and demonstrated by Boger in 1984 (Scheme 1) [3]. “Divergent” is defined as a common intermediate (preferably an advanced intermediate) being converted, separately, to at least two natural products. Applications of divergent synthesis include not only preparing molecules in the same family but also accessing natural products that have the same molecular skeletons from different families. Divergent total synthesis, also defined as “collective total synthesis” by MacMillan, is when multiple skeletons of natural products are prepared from a versatile common intermediate [4].

Figure 1.
Schematic explanation of linear and divergent synthesis.

Scheme 1.
Boger’s divergent total syntheses of rufescine and imetuteine.
Compared with linear synthesis that can only achieve one natural product at a time, the divergent strategy provides a more efficient approach to access many natural products with similar or different molecular skeletons from a versatile molecule through a number of routine chemical operations. In addition, the divergent synthesis of natural products is also more conducive to building a natural-product-like compound library to conduct more biological activity screening and innovative drug development based on the bioactive natural products.
The application of both strategies requires organic chemists to be familiar with the skeleton, functional group, oxidation state, and other characteristics of the target molecules. One of the most challenging steps is to design a suitable advanced intermediate that can be easily prepared and quickly converted to as many target molecules as possible. Meanwhile, the common intermediate should also be close to the target molecule to reduce the total number of synthesis steps.
2. Selected Deconstructive and Divergent Syntheses of Natural Product (2021–2023)
In 2021, the Reisman group from the California Institute of Technology reported the divergent total syntheses of three C19 diterpenoid alkaloids: (−)-talatisamine, (−)-liljestrandisine, and (−)-liljestrandinine from phenol (Scheme 2a) [5][13]. The highlights of this work include (1) a 1,2-addition/semipinacol rearrangement sequence to efficiently couple two complex fragments and construct the quaternary carbon center and (2) an intramolecular aziridination and radical cyclization to assemble the pentacyclic skeleton of the target alkaloids.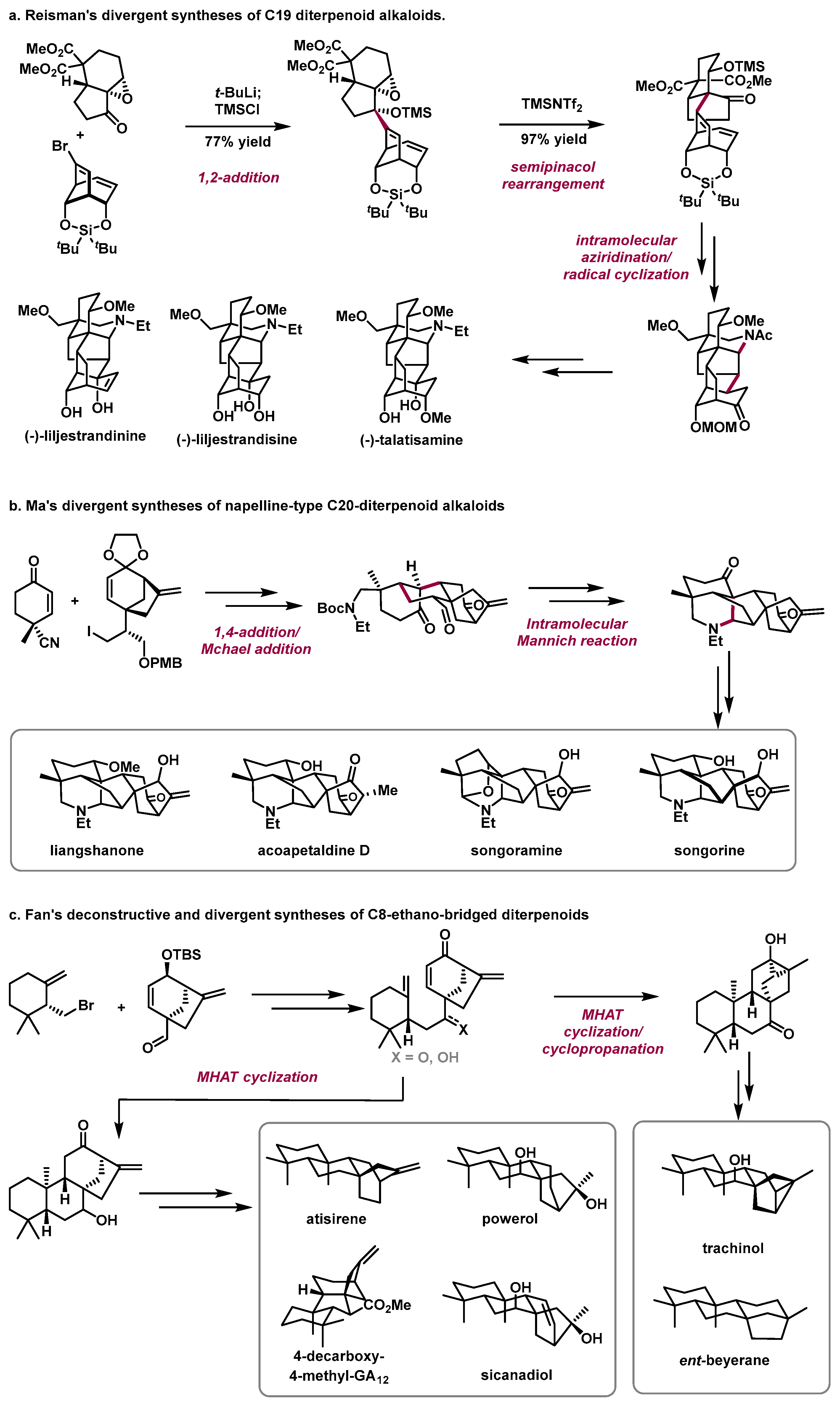
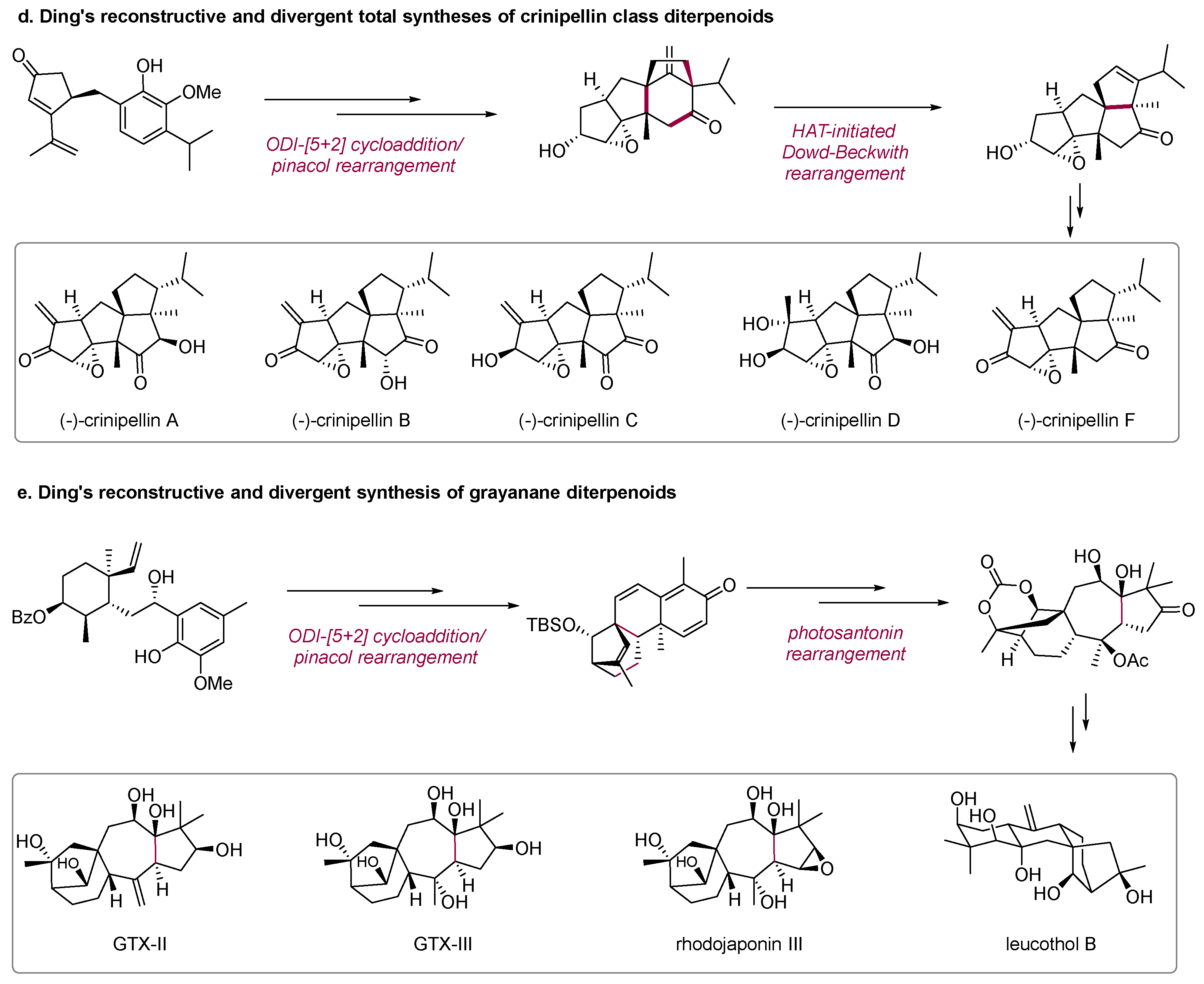


Scheme 2.
Selected examples of deconstructive and divergent syntheses of natural products.
3. Divergent Syntheses of Fawcettimine-Class Lycopodium Alkaloids
Lycopodium alkaloids are structurally complex natural products with quinolizine, pyridine, or α-pyridone, originally identified in the Lycopodium genus. Lycopodium alkaloids exhibit important biological activities. For example, Huperzine A is a potent inhibitor of acetylcholinesterase (AChE) and shows promise in the treatment of Alzheimer’s disease (AD). The fawcettimine-class Lycopodium alkaloids are a class of structurally unique alkaloids with complex and unique skeletons bearing quaternary carbon centers, such as fawcettimine (1); fawcettidine (2); lycojaponicumins C (3); and 8-deoxyserratinine (4) (Scheme 3) [10][18]. In particular, lycojaponicumins C (3), isolated by Yu and co-workers in 2012 from the traditional Chinese medicine Lycopodium japonicum, exhibits lipopolysaccharide (LPS)-induced pro-inflammatory factors in BV2 macrophages [11][19]. The fawcettimine-class Lycopodium alkaloids feature fused tetracyclic skeletons, including two common cis-hydroindene (6/5 bicycle) motifs and two diverse ring systems bearing quaternary carbon centers. The Lycopodium alkaloids have attracted great attention from synthetic chemists and medicinal chemists for their unique chemical structures and broad biological activities. During the past decade, several elegant approaches for the total synthesis of Lycopodium alkaloids have been reported [12][20].
Scheme 3.
Structures of representative fawcettimine-class
Lycopodium
alkaloids.

Scheme 4.
Synthesis of tetracyclic azide (
13)
.

Scheme 5. Divergent syntheses of fawcettimine-class Lycopodium alkaloids fawcettimine (1), fawcettidine (2), lycojaponicumins C (3), and 8-deoxyserratinine (4).
4. Deconstructive Syntheses of Cyclopiane Class Tetracyclic Diterpenes
The Cyclopiane class tetracyclic diterpenes are a class of unique bioactive natural products isolated from fermentation of marine-derived entophytic fungi of the Penicillium genus [15][16][17][23,24,25]. Conidiogenone (19) and conidiogenol (20) exhibit potent conidiation-inducing activity, while conidiogenone B (21) shows high antibacterial activities (Figure 2). The structures of the Cyclopiane class tetracyclic diterpenes feature a highly fused and strained tetracyclic (6/5/5/5) skeleton, 6–8 consecutive chiral centers, and four sterically hindered quaternary carbon centers. The Cyclopiane-class tetracyclic diterpenes have attracted great attention for their unprecedented chemical structures and important biological activities. During the past decade, several elegant approaches for the total syntheses of Cyclopiane class tetracyclic diterpenes have been reported [18][19][20][21][26,27,28,29]. The first total syntheses of three Cyclopiane class tetracyclic diterpenes, namely, conidiogenone (19), conidiogenol (20), and conidiogenone B (21), was accomplished by the Tu group in 2016 through the use of a well-designed semipinacol rearrangement as a key step to construct the requisite spirocyclic (6/5) skeleton and sterically hindered quaternary carbon center of the target molecules [18][26].
Figure 2.
Representative bioactive
Cyclopiane
-class tetracyclic diterpenes.

Scheme 6.
Deconstructive syntheses of the
Cyclopiane
-class tetracyclic diterpenes (
19
–
21
).
5. Asymmetric Total Syntheses of Serrulatane and Amphilectane Diterpenoids
Serrulatane and amphilectane diterpenoids are a class of polycyclic natural products that exhibit broad biological activities, such as anti-malarial, anti-tuberculosis, and anti-bacterial properties. (Figure 3). (−)-Microthecaline A (37) is a unique nitrogen-containing serrulatane-type diterpenoid that exhibits anti-malarial activity against Plasmodium falciparum (IC50 = 7.7 μM) [22][30]. (−)-Leubethanol (38) is a serrulatane diterpene isolated from the roots of Leucophyllum frutescens with important anti-tuberculosis activity (MIC 6.25–12.50 μg/mL) [23][31]. Because of their wide-ranging biological activities and challenging structures, synthesis of serrulatane and amphilectane diterpenoids have attracted great attention from synthetic chemists, and several total syntheses have been accomplished in the past decade. In 2020, the Dong group developed a divergent and innovative approach to quickly access diverse serrulatane diterpenoids, namely, (−)-microthecaline A (37), (−)-leubethanol (38), (+)-seco-pseudopteroxazole (39), amphilectane diterpenoids (+)-pseudopteroxazole (40), (+)-pseudopterosin G–J aglycone (41), and (−)-pseudopterosin A–F aglycone (42), by using a Rhodium catalytic C–C/C–H cascade reaction to construct the common skeleton, α-tetralone [24][32].
Figure 3.
Representative of bioactive serrulatane and amphilectane diterpenoids.

Scheme 7.
Total synthesis of (−)-microthecaline A (
37
) and (−)-leubethanol (
38
).

Scheme 8. Total syntheses of terpenoids (+)-seco-pseudopteroxazole (39), (+)-pseudopteroxazole (40), (+)-pseudopterosin G–J aglycone (41), and (−)-pseudopterosin A–F aglycone (42).
6. Deconstructive Synthesis of Morphine Alkaloid (−)-Thebainone A
Morphine and codeine have attracted great attention for their powerful biological activity and medical applications (Figure 4). Modification of the morphine alkaloids is still an active field in drug discovery. Therefore, developing a new asymmetric synthetic route of morphine alkaloids and their analogues is highly desirable for exploring their potential utilities in drug discovery and development. In 2021, the Dong group reported a concise enantioselective deconstructive synthesis of the morphine alkaloid thebainone A for the first time, as well as formal synthesis of codeine and morphine from commercially available materials [2][25][2,33]. The high efficiency of the synthetic strategy is enabled by an asymmetric Rh-catalyzed C–C activation reaction (cut-and-sew) to access the all-carbon fused-rings structure and quaternary carbon center.
Figure 4.
Morphine and related alkaloids.

Scheme 9.
Deconstructive synthesis of morphine alkaloids (−)-thebainone A (
67
).