Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Zijian Zhu and Version 2 by Dean Liu.
Breast cancer continues to pose a significant healthcare challenge worldwide for its inherent molecular heterogeneity.
- breast cancer
- heterogeneity
- single-cell genome
1. Introduction
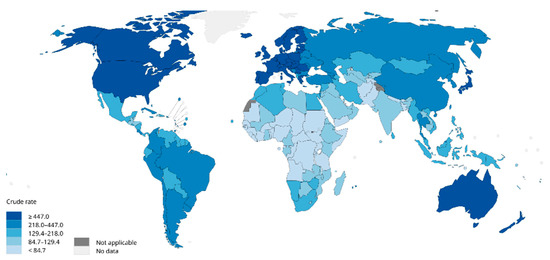
Breast cancer is one of the most prevalent cancers in women globally. In 2012, there were 464,000 diagnosed cases of breast cancer and 131,000 deaths among European women [1]. In 2020, breast cancer accounted for 2.26 million new cases globally, surpassing lung cancer (2.2 million cases) to become the most commonly diagnosed cancer worldwide (Figure 1). In the same year, the disease caused an estimated 685,000 deaths [1][2][3][1,2,3]. Projections suggest that by 2030, new cases will reach 3.9 million globally, with fatalities rising to 766,000 [4].
Despite considerable strides in both laboratory research and the clinical practice of breast cancer, the global incidence and mortality rates continue to rise. At the root of this persisting issue is the heterogeneous nature of breast cancer, which is not a monolithic disease, but a spectrum of distinct subtypes, each representing a unique malignancy within the breast’s cellular makeup. Current research categorizes these molecular subtypes into six major classes: (i) hormone-receptor-positive breast cancer (ER+), (ii) hormone receptor/HER2-positive breast cancer (ER+/HER2+), (iii) HER2-positive breast cancer (HER2+), (iv) basal-like breast cancer, (v) claudin-low subtype, and (vi) normal-like subtype [5]. The distinct subtypes of breast cancer each present unique clinical features and associated risk factors. This diversity extends to treatment responses and long-term patient survival, which differ significantly across the subtypes. This inherent complexity adds layers of challenge to the effective diagnosis and treatment of breast cancer. For instance, the basal-like subtype of breast cancer, characterized by high rates of cellular proliferation, is associated with distinct risk factors, which include the early onset of menstruation, a younger age at the first full-term pregnancy, and the accumulation of abdominal fat. In contrast, patients with the claudin-low subtype, marked by an enrichment of epithelial–mesenchymal transition markers, typically exhibit pronounced invasiveness. Such individuals often bear the burden of exposure to chemicals and radiation in their early years, leading to a high load of DNA damage induced by cancer genes and early chromosomal instability (CIN) [6].
The inherent heterogeneity of breast cancer presents considerable hurdles for conventional diagnostic and therapeutic approaches. Generally, traditional methods depend on analyzing bulk tumor tissue samples, a process which, by considering the average expression levels, may obscure the underlying heterogeneity, complicating accurate tumor classification. But emerging technologies such as single-cell analysis techniques offer promising alternatives, already being widely used in oncology research. These techniques, by investigating gene expression, phenotypes, protein levels, and other cellular properties at an individual cell level, are well suited to address the challenge of tumor heterogeneity (Figure 2) [7][8][9][7,8,9]. Particularly for highly heterogeneous cancers like breast cancer, a single-cell analysis can help to predict cellular evolution during tumor progression. The analysis of genetic and epigenetic variations as well as gene expression at the single-cell level is among the techniques that enhance the precision of predicting tumor development trends, evaluating treatment outcomes, and forecasting patient prognosis. Furthermore, single-cell analysis techniques play a crucial role in devising novel therapeutic strategies. These methods allow for the examination of genetic variations and phenotypic characteristics of tumor cells in detail, which can lead to the identification of new therapeutic targets. Consequently, this paves the way for the development of highly targeted, precise treatment strategies, enhancing the ability to predict treatment efficacy and potential drug resistance.
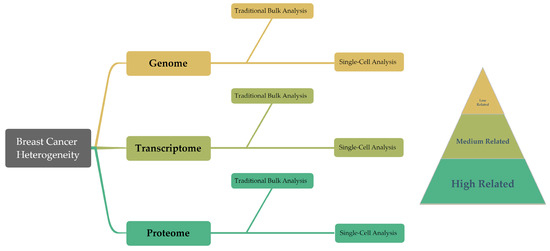
Figure 2. This diagram presents an overview of the structure of the article, summarizing the traditional bulk techniques and single-cell findings in genomics, transcriptomics, and proteomics. Moreover, these three groups demonstrate a progressive correlation with cancer cell behavior in the research on breast cancer heterogeneity.
Single-cell gene sequencing, leveraging the power of next-generation sequencing (NGS), has emerged as a pivotal tool for investigating breast cancer heterogeneity [10]. Unlike traditional Sanger sequencing, NGS systems use massive parallel sequencing to yield billions of DNA reads, from 36 to 150 base pairs, which can be aligned to the human genome. This alignment allows for the detection of various genetic variations, including single-nucleotide mutations, small insertions/deletions, and copy number variations, offering a comprehensive view for streamlining the development of targeted treatment strategies. A case in point is HER2-positive breast cancer, where single-cell sequencing detects the diversity in HER2 gene amplification across different cells, facilitating the formulation of bespoke treatment plans [11]. The versatile utility of NGS extends to RNA sequencing (RNA-seq), which is a high-throughput technique enabling quantitative and sequence analyses of diverse RNA types, along with their expression levels in cells and tissues. RNA-seq facilitates an in-depth exploration of gene expression regulation, signaling, and metabolic pathways pertinent to breast cancer, thereby enriching theour understanding of its molecular mechanisms. Intriguingly, beyond the recognized impact of non-coding RNAs on breast cancer progression, even the half-life of mRNA serves as an informative marker [12].
In molecular profiling, the strength of the correlation between molecular patterns and cellular behavior is pivotal, as a higher correlation implies a more accurate reflection of the tumor’s actual condition [13]. Complementing single-cell genomics and transcriptome, the emergence of single-cell proteomics offers another potent instrument for investigating breast cancer heterogeneity. This technique enables the detection and analysis of protein expression at the single-cell level. It provides a more precise view of protein expression compared to its RNA-sequencing counterpart, revealing insights into protein localization and intra-cellular interactions. For instance, immunohistochemistry provides a more direct appraisal of a patient’s tumor condition. This technique plays a pivotal role in breast cancer diagnosis, as it involves staining clinical samples of breast cancer tissues to reveal the expression of crucial proteins, including ER, PR, and HER2. The resultant immuno-stained samples offer a visual map for clinicians, aiding them in identifying the subtype of breast cancer and enabling effective pathological staging and treatment selection. Through techniques like immunohistochemistry, reswear can not only discern between these subtypes, but also detect signs of lymph node metastasis and monitor potential tumor recurrence. Such insights are pivotal for guiding treatment decisions and tracking the progression of the disease [14][15][16][14,15,16].
2. Genomic Profiling
2.1. Traditional Genomic Profiling
In 1994, the BRCA1 gene was identified through positional cloning, followed by the discovery of the BRCA2 gene in 1995. These genes play a crucial role in DNA damage repair and maintaining genomic stability, thereby reducing the risk of tumor development [17][18][17,18]. BRCA1 and BRCA2 are tumor suppressor genes involved in repairing dsDNA breaks. Mutations in these genes significantly increase the lifetime risk of developing breast cancer. Inherited mutations in BRCA1 and BRCA2 account for a small percentage of breast cancer cases. Tumors associated with BRCA1 mutations often exhibit a basal-like phenotype and a higher histological grade, while those linked to BRCA2 mutations resemble sporadic tumors more closely. Several specific scenarios can notably elevate the incidence of breast cancer: (1) sequence variants encoding premature termination codons such as nonsense or frameshift mutations occurring prior to the 1855th amino acid in BRCA1 and the 3309th amino acid in BRCA2; (2) mutations located at splice site consensus sequences—either the first or second base positioned upstream or downstream of an exon; (3) copy number loss mutations leading to frameshift mutations prior to the 1855th amino acid of BRCA1 and the 3309th amino acid of BRCA2, or mutations eliminating one or more exons not predicted or confirmed to produce functional in-frame RNA isoforms capable of restoring BRCA1/2 gene function; and (4) copy number repeat variations of any size resulting in the duplication of one or more exons, and proven to cause frameshift mutations before the 1855th amino acid of BRCA1 and the 3309th amino acid of BRCA2 [17][19][17,19]. Individuals harboring potential pathogenic variants in the BRCA1/2 genes can benefit significantly from timely education and early screening. According to the European Society for Medical Oncology (ESMO) guidelines [20], females identified with having mutations in BRCA1, BRCA2, or other high-penetrance genes should initiate breast cancer prevention education from the age of 18, maintain vigilant awareness of breast conditions, and comply with regular medical check-ups. Physicians advocate for annual clinical breast examinations complemented by breast X-ray imaging and magnetic resonance imaging (MRI) assessments starting from the age of 25 [21]. Despite the insights gained from early genetic knowledge, they did not immediately translate into clinical treatment strategies. The initial clinical data highlighted that BRCA-associated tumors exhibited high sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors. These inhibitors act on the PARP-mediated DNA damage repair mechanism, thereby disrupting the tumor’s ability to repair its DNA. As of now, PARP inhibitors are primarily accessible through clinical trials [22]. With the deepening understanding of breast cancer and the widespread use of NGS, more relevant genes have been discovered (Table 1).Table 1. Breast-cancer-relevant gene, discovery year, involved process, and mutation risk.
| Gene | Discovery | Involved Process | Mutation Risk | Reference |
|---|---|---|---|---|
| PTEN | 1997 | apoptosis, cell cycle, and signal transduction | activation of proliferation and survival signals | [23] |
| STK11 | 1997 | cell cycle, metabolism, and energy balance | activation of cell proliferation and metabolic pathways | [24] |
| CHEK2 | 1999 | DNA repair and cell apoptosis | impairments in DNA repair and cell apoptosis processes | [25] |
| PIK3CA | 2004 | regulation of signaling pathway | activation of survival signals | [26] |
| AKT1 | 2007 | regulation of signaling pathway | activation of cell proliferation and survival signals | [27] |
| BARD1 | 2010 | DNA repair and cell apoptosis | increased susceptibility to breast cancer | [28] |
| NF1 | 2015 | regulation of signaling pathway | increased rate of developing breast cancer | [29] |