Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Bong-Woo Lee.
Psoriatic arthritis (PsA) is a persistent, inflammatory disease that affects individuals with psoriasis, arthritis, and enthesitis. Inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-23 (IL-23), and interleukin-17 (IL-17) play a pivotal role in both the onset and progression of PsA. These cytokines are generated by activated immune cells and stimulate the attraction of inflammatory cells to the synovium and joint tissues, resulting in the deterioration of cartilage and bone.
- psoriatic arthritis
- inflammatory cytokines
- tumor necrosis factor-alpha
1. Introduction
Psoriatic arthritis (PsA) is a persistent condition associated with immune-related inflammation that affects joints and other elements of the musculoskeletal system, in addition to skin manifestations. It is estimated that this disease affects approximately 1–2% of the worldwide population [1]. PsA is recognized as part of psoriatic diseases and belongs to a complex group of spondyloarthritides (SpA) [2,3,4][2][3][4].
PsA is identified by a varied range of clinical manifestations, such as peripheral joint inflammation, inflammatory back pain, uveitis, enthesitis, tenosynovitis, psoriasis, and nail changes [5]. Historically, PsA was regarded as a relatively benign condition. However, it is now widely acknowledged that the functional impact of PsA is comparable to that of other types of inflammatory arthritis, including rheumatoid arthritis (RA) and axial SpA (axSpA) [1]. In many individuals with PsA, the disease exhibits characteristics of progressive and destructive changes. Significant pathological changes occur already in the early stages of PsA, with approximately half of the patients displaying structural damage within 2 years from disease onset [6]. Research has demonstrated that a delay in diagnosing PsA is linked to unfavorable outcomes impacting the quality of life [7].
While the precise cause of PsA is not fully comprehended, it is believed to arise from a complex interplay of environmental, genetic, and immunological factors [2]. Specific human leukocyte antigen (HLA) alleles, namely HLA-B*27, HLA-B*08, HLA-B*38, and HLA-B*39, exhibit a strong correlation with the prevalence of PsA [8]. It is hypothesized that these alleles may render individuals more susceptible to PsA by modifying the immune response to self-antigens. Furthermore, environmental factors, including infections and mechanical stress, can potentially act as triggers for PsA development in individuals with a genetic predisposition [9,10][9][10]. The microbiome of the gut and skin might have an impact on the development of PsA [11,12][11][12]. Immunological factors, including cytokines and chemokines, are believed to have a crucial role in PsA pathogenesis. Tumor necrosis factor-alpha (TNF-α), interleukin-17 (IL-17), and interleukin-23 (IL-23) have been recognized as significant contributors to the inflammatory response, tissue damage, and bone erosion associated with PsA. Therapies targeting these cytokines have been developed and used as treatments for patients with PsA. Despite advancements in comprehending the molecular mechanisms that cause PsA, the existing knowledge is not sufficient to meet significant clinical needs in effective treatments [13]. Overall, PsA pathogenesis is complex and multifactorial, involving genetic, environmental, and immunological factors.
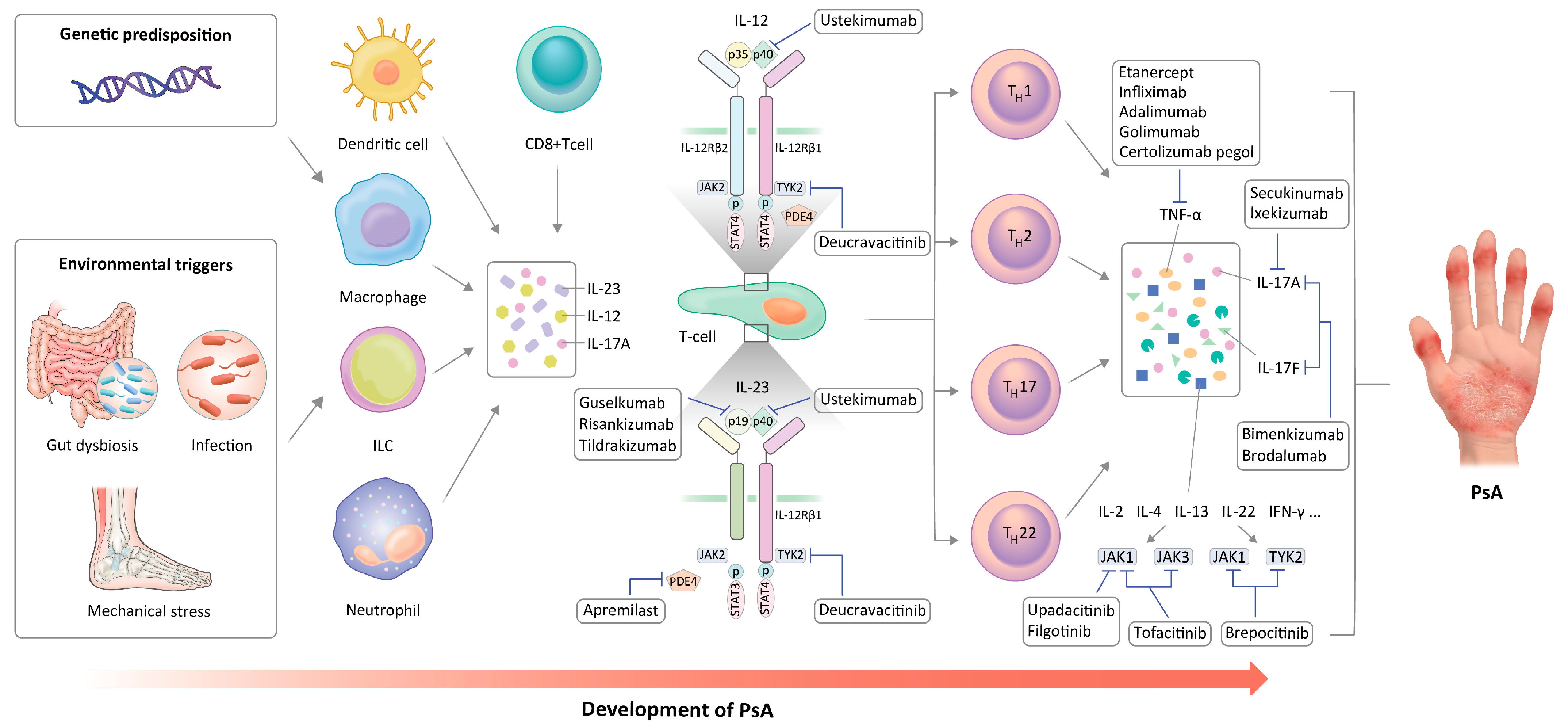
2. Immune Cells in PsA Pathogenesis
PsA is a chronic and complex condition characterized by the involvement of the skin, enthesis, and synovium. The degree of this involvement may vary among individuals and is influenced by genetic and environmental factors [14]. PsA is initiated by the stimulation of dendritic cells (DCs) and macrophages. These cells expose antigens to T cells through the toll-like receptor-2 (TLR-2) signaling pathway, facilitated by major histocompatibility complex (MHC) I. This activation occurs in response to a combination of genetic predisposition, environmental conditions, and biophysical factors. Consequently, the activation of both innate and adaptive immune responses leads to the increased secretion of various cytokines, such as IL-1, IL-6, TNF-α, IL-17, and IL-23 [15]. In addition, the activation and invasion of T cells and macrophages are considered essential in the initiation of inflammatory and destructive processes in the joints [16]. Specifically, DCs induce T cell differentiation by presenting antigens and secreting various pro-inflammatory cytokines. Increased expression of TLR-2 has been detected in the immature DCs of PsA patients [17]. Patients with PsA exhibit an elevated proportion of immature myeloid DCs compared to plasmacytoid DCs in their synovial fluid [18]. In PsA, TLR stimulation leads to the polarization of T cells towards the Th1 subset, subsequently increasing the production of TNFα, IFN-γ, and IL-2 [17]. Plasmacytoid DCs generate cytokines, including IFN-γ, TNF-α, IL-12, and IL-23, which subsequently serve as signals for the clonal proliferation of CD4+ and CD8+ T cells [19]. Activated macrophages participate in various pro-inflammatory processes within the synovium. A reduction in the quantity of CD68+ macrophages was observed in the synovium of patients with PsA who responded to treatment, which highlighted the role of these cells in PsA pathogenesis [20]. Mast cells located in the synovium induce angiogenesis, neutrophil recruitment, and proliferation of synovial fibroblasts, indicating that these cells may actively contribute to the inflammatory arthritis [24][21]. In the synovium of peripheral SpA, mast cells are the predominant source of IL-17A, whereas only a small number of IL-17-positive T cells was identified [25][22]. Likewise, tissue-resident mast cells may increase inflammation by producing IL-17A. However, it remains to be established in more detail how the release of IL-17A from mast cells is controlled in the affected tissue during inflammation [26][23]. Other various cells in the synovial tissue could potentially participate in PsA pathogenesis. Notably, elevated levels of type 3 innate lymphoid cells (ILCs), which produce IL-17 and IL-22, have been observed in the synovial fluid of PsA patients [27][24]. Conversely, there was a decrease in the number of type 2 ILCs, which typically generate IL-4, IL-5, IL-9, and IL-13. Significantly, the ratio of type 2 ILCs to type 3 ILCs showed a strong correlation with both clinical indicators of disease activity and the radiographic evidence of arthritis and bony destruction. In addition, it has been shown that environmental stress signals activate natural killer (NK) cells, leading to their involvement in the harmful processes associated with PsA development [28,29][25][26]. In the development of PsA, innate immune cells contribute by producing IL-12 and IL-23, which stimulate the differentiation of T cells into distinct subtypes referred to as Th1 and Th17 helper T cells [5,31][5][27]. Consequently, IL-22 and IL-17 are secreted, which in turn increase the secretion of TNF-α [32][28]. T cells are the primary contributors to PsA pathogenesis, as evidenced by the elevation of CD4+ T17 cells that secrete IL-23 and IL-17 in both the peripheral blood and synovial fluid [33,34,35][29][30][31].3. Key Inflammatory Cytokines in PsA
3.1. Role of TNF-α in PsA
TNF-α, a pro-inflammatory cytokine produced by cells like macrophages and monocytes, is prominently present during inflammatory processes [42][32]. It serves as a key component of pro-inflammatory immune responses. TNF-α interacts with two distinct receptors, TNFR1 and TNFR2, initiating separate signaling pathways that trigger apoptosis, cell differentiation, proliferation, and migration. As a result, TNF-α contributes to the development of inflammatory reactions. Additionally, TNF-α plays a crucial role in promoting bone resorption by directly activating osteoclast precursor cells via the RANKL signaling pathway [43][33]. At high concentrations, TNF-α causes adverse effects, including uncontrolled inflammatory responses, increased formation of osteoclast precursors, and enhanced osteoclast activity, ultimately resulting in bone resorption [44][34]. TNF-α plays a pivotal role in the pathogenesis of SpA manifestations, encompassing skin, enthesis, joint, and spine involvement, which can be considered as the secondary effects of hyperactivated TNFα-dependent inflammation due to the activation of both innate and adaptive immune responses [45][35]. The evidence supporting the involvement of TNF-α in inflammatory forms of arthritis is particularly compelling [43][33]. Elevated levels of TNF-α are detected in the synovial fluid and tissues of patients with RA and PsA. TNF-α is recognized as a key contributor to the initiation and persistence of joint inflammation and damage in PsA. It facilitates the production of enzymes that degrade the cartilage and bone, thereby leading to joint destruction. Moreover, TNF-α induces angiogenesis, which further fuels the progression of joint inflammation [2].3.2. Role of IL-23 in PsA
IL-23 is a cytokine composed of two subunits, p19 and p40, with the latter being shared with IL-12. Its main function is to facilitate the activation, differentiation, and proliferation of Th17 cells, which are responsible for producing effector cytokines like IL-17A and IL-22. However, it is worth noting that IL-17 production can also occur independently of IL-23 [48][36]. The injection of IL-23 causes a skin disease like psoriasis in mice with normal expression of IL-17, while IL-17 knockout mice do not exhibit such disease manifestations. The physiological processes governed by the IL-23 signaling strongly support its crucial role in maintaining the homeostasis of the intestinal barrier. Intestinal inflammation is linked to the increased permeability of the gut barrier, which facilitates the infiltration of pathogen-associated molecular patterns into the host system. The compromised intestinal barrier leads to a persistent activation of the host’s defense mechanisms, which subsequently triggers an excessive production of IL-23, the cytokine that plays a pivotal role in the development of SpA pathogenesis [50][37]. IL-23 demonstrates a remarkable capacity to induce inflammation in various anatomical sites, including the gut, skin, enthesis, and joints, with high efficiency. Notably, IL-23 alone could directly induce the characteristic clinical features of psoriasis and PsA in mice. Experimental evidence has shown that IL-23 expression in mice results in the emergence of enthesitis, osteitis, new bone formation, and bony erosion [51][38].3.3. Role of IL-17 in PsA
IL-17, a group of pro-inflammatory cytokines comprising six known members (IL-17 A/B/C/D/E/F), is mainly generated by Th17 T cells, and its presence has been associated with the development of psoriatic skin lesions and enthesitis [55][39]. IL-17 is secreted by multiple cell populations, including Th17 helper cells, γ/δ T cells, interstitial lymphoid cells, neutrophils, and mast cells [2]. Several studies have suggested that a combination of cytokines, including IL-23, TGF-β, IL-6, IL-1β, and IL-21, can induce the differentiation of naïve T cells into Th17 cells [56][40]. IL-23 stimulates activated Th17 cells, leading to the release of IL-17. While many members of the IL-17 family contribute to inflammation and participate in immune-related diseases and defense against pathogens, they also have various functions in preserving mucosal integrity, responding to allergens, supporting anti-inflammatory responses, and regulating the function of lymphocytes [57][41]. Among these members, IL-17A and IL-17F are particularly significant in PsA. IL-17A and IL-17F have the ability to form both homo- and heterodimers, which can bind to the IL-17 receptor and stimulate signaling pathways downstream. This activation leads to the release of pro-inflammatory cytokines. In PsA, IL-17A is considered the main pro-inflammatory cytokine, although IL-17F likely also has some effects [58][42]. IL-17A has a multitude of effects on diverse immune cells in the skin and joints, stimulating inflammation, coagulation, and causing damage to bones and joints. IL-17A exerts significant biological effects by attracting neutrophils, Th17 cells, and ILC3s to the affected tissues, particularly the skin and entheses. This recruitment process contributes to the generation of the immune response [50][37]. IL-17A acts as a mediator that aggravates tendon inflammation by initiating neutrophil responses. This inflammatory process clinically presents as enthesitis and tendinitis, which are characteristic features of PsA [59][43].3.4. Other Cytokines or Chemokines in PsA
While the therapeutic efficacy of apremilast, a phosphodiesterase 4 (PDE4) inhibitor, in PsA is linked to its capacity to suppress the production of TNF-α, IL-17, and IL-23, the effectiveness of Janus kinase (JAK) inhibition in PsA treatment suggests the participation of other cytokines in the inflammatory mechanisms underlying this condition [62,63][44][45]. IL-22 is a cytokine generated by Th22 cells, a recently identified subset of T helper cells expressing chemokine receptor (CCR)10. Serum levels of IL-22 are notably elevated in patients with PsA compared to those in patients with psoriasis only [64][46]. In regulating bone homeostasis, IL-22 has been shown to have dual effects on both osteoclasts and osteoblasts. It has been reported to promote the expression of RANKL and upregulate osteoclastogenesis [65][47]. On the other hand, IL-22 promotes osteoblast differentiation and function, leading to enhanced bone formation. It can stimulate the production of various osteogenic factors and extracellular matrix proteins by osteoblasts, thereby supporting bone mineralization and overall bone formation [66][48]. IL-32 activates monocytes, promoting their differentiation into macrophages or DC, and also triggers the production of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-8 through the inflammatory signaling pathway involving NF-κB and p38 mitogen-activated protein kinase [67][49]. Plasma levels of IL-32 are elevated in patients with PsA compared to those in healthy controls, and notably higher than in patients with psoriasis alone. These findings indicate that plasma IL-32 levels in patients with psoriasis only increase prior to PsA onset, and further elevate upon PsA aggravation [68][50]. IL-33, a member of the IL-1 family, interacts with the ST2 receptor and activates various immune cells, including ILC2 and Th2 cells. Furthermore, the IL-33/ST2 axis appears to play a role in regulating the Th17 response [69][51]. Prior to treatment, patients with PsA exhibit a notably higher expression of CXCL12 compared to that in patients with psoriasis vulgaris [73][52]. The significant up-regulation of myeloid cells in PsA has generated increased attention from researchers towards myeloid-derived pro-inflammatory mediators, including osteopontin and CCL2, found in the synovial fluid of patients with PsA [74][53]. Elevated expression levels of the CCL1, CCL20, CXCL5, and CX3CL1 genes were detected in both the synovial fluid and peripheral blood cells of individuals with PsA [75][54].4. Cytokine-Targeted Therapies in PsA
4.1. TNF-α Inhibitors in PsA
TNFi, a class of pharmaceuticals that antagonize TNF-α activity, have shown efficacy in the management of PsA. These medications, including etanercept, certolizumab pegol, infliximab, adalimumab, and golimumab, decrease joint inflammation, improve physical function, and prevent joint damage in patients with PsA [79][55]. TNFi have shown significant efficacy in treating all SpA manifestations and are recommended by the GRAPPA and EULAR for the management of enthesitis and dactylitis [80][56]. Among TNFi, etanercept was less effective than TNFi representing monoclonal antibodies in the treatment of uveitis or inflammatory bowel disease, which are extra-musculoskeletal manifestations of PsA [81,82][57][58]. On the other hand, etanercept exhibits lower immunogenicity compared to other TNFi and does not necessitate concurrent methotrexate administration to maintain its long-term efficacy. In a randomized, controlled phase III trial, both etanercept monotherapy and combination therapy with etanercept and methotrexate had superior efficacy compared to that achieved with methotrexate monotherapy in patients with PsA. Furthermore, the addition of methotrexate did not synergistically enhance the efficacy of etanercept [83][59].4.2. IL-17 Inhibitors in Psoriatic Arthritis
The prominent involvement of IL-17 in the immune-related mechanism of PsA validates the use of IL-17 inhibitors for all forms of psoriatic disease. Secukinumab became the first IL-17 inhibitor to receive approval for the treatment of PsA. After that, ixekizumab was approved, and bimekizumab and brodalumab are also being studied for their potential efficacy in PsA. Secukinumab is a human IgG1k class monoclonal antibody that specifically targets IL-17A. The FUTURE trials have shown its superior efficacy over placebo in multiple domains of PsA, including spondylitis, peripheral arthritis, enthesitis, dactylitis, psoriasis, and nail disease [85,86][60][61]. Ixekizumab is a IgG4κ subclass monoclonal antibody, produced using recombinant technology, which specifically targets IL-17A. In both TNFi refractory and naïve patients, ixekizumab exhibited superiority over the placebo in terms of ACR responses, inhibition of radiographic progression, and various clinical aspects, similar to the effects observed with secukinumab [88,89][62][63]. Ixekizumab was superior to adalimumab in achieving the complete remission of psoriasis, as indicated by the psoriasis area and severity index (PASI)100 response at week 24 in a head-to-head clinical trial. Bimekizumab, a humanized IgG1κ monoclonal antibody targeting IL-17A and IL-17F, demonstrated notable improvements in the ACR50 response compared to the placebo in PsA patients treated for 12 weeks, as evidenced by data from the randomized controlled trial (BE ACTIVE). Moreover, sustained improvements in pain and fatigue were observed over a period of 3 years [91,92][64][65]. Brodalumab, a human IgG2 antibody, specifically binds to IL-17 receptor A, leading to the inhibition of IL-17A, IL-17F, and IL-17E. In multicenter phase III clinical trials (AMVISION-1 and 2), brodalumab demonstrated a superior ACR response compared to the placebo group at week 24. Additionally, significant improvements in dactylitis, enthesitis, and psoriasis resolution rates were observed in the brodalumab treatment group [94][66]. IL-17 inhibitors are recommended for the management of most manifestations in PsA. A recent meta-analysis showed similar effectiveness of anti-IL-17A agents and TNFi in the treatment of arthritis, with superior cutaneous responses to anti-IL-17A agents [95][67].4.3. IL-23 Inhibitors in PsA
The first IL-23 inhibitor introduced to the market was ustekinumab, which reduces the activities of both IL-23 and IL-12 by specifically targeting their shared p40 subunit. Furthermore, monoclonal antibodies guselkumab, risankizumab, and tildrakizumab specifically target the p19 subunit of IL-23 [98][68]. Ustekinumab effectively inhibits the differentiation of Th1 and Th17 cells. Phase 3 clinical trials (PSUMMIT trials 1 and 2) showed that this drug effectively treated skin manifestations, peripheral arthritis, enthesitis, and dactylitis in both TNFi-experienced and TNFi-naïve patients with PsA [15]. The use of ustekinumab resulted in a higher rate of achieving the primary outcome, defined as an ACR20 response at week 24, compared to the placebo group [99][69]. Guselkumab, a monoclonal antibody that specifically binds to the p19 subunit and blocks the activity of IL-23, has obtained approval for the treatment of PsA. In clinical trials (DISCOVER 1&2), the ACR20 response to guselkumab was significantly higher than to the placebo at week 24 both in TNFi-naïve and TNFi-treated PsA patients [105,106,107][70][71][72]. The safety and clinical improvements were consistently maintained over 2 years [108][73]. Guselkumab rapidly decreased the levels of type 17 effector cytokines as early as week 4 after the start of the treatment. Risankizumab, a monoclonal antibody targeting IL-23, has received the U.S Food and Drug Administration (FDA) approval for the treatment of PsA. The clinical trials conducted on both bDMARD-naïve and bDMARD-experienced patients with PsA showed that significantly more patients achieved an ACR20 response at week 24 after the treatment with risankizumab compared to the effects of the placebo [110,111][74][75].4.4. JAKi in PsA
The Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway plays a crucial role in the activation of the immune system in response to cytokines and growth factors, facilitating the transmission of signals from cell-membrane receptors to the nucleus. This pathway involves four JAK proteins, namely JAK1, JAK2, JAK3, and tyrosine-protein kinase 2 (TYK2). These molecules intricately interact with different members of the signal transducers and activators of the transcription (STAT) family, ultimately regulating the transcription of genes downstream of the pathway [114][76]. Tofacitinib is a small molecule that selectively blocks the activities of JAK1 and JAK3. In two separate phase III trials with a randomized, double-blind design for PsA patients, ACR20 response rates and improvement in the health assessment questionnaire–disability index scores were significantly greater after tofacitinib than after the placebo [115,116][77][78]. The tofacitinib group showed improved clinical outcomes of peripheral arthritis, dactylitis, enthesitis, and psoriasis compared to the outcomes in the placebo group during the first 6 months of therapy [117][79]. Upadacitinib, a highly specific inhibitor of JAK1, has recently been approved as the latest drug in its class for the treatment of PsA. The phase 3 clinical trial SELECT-PsA 1 provided evidence of the effectiveness of upadacitinib in achieving a significant ACR20 response when compared to the effects of placebo. Significant improvements in secondary endpoints, such as decreased radiographic progression, attainment of minimal disease activity, resolution of dactylitis, and improvement in enthesitis, were observed in both the 15 and 30 mg upadacitinib treatment groups, surpassing the results seen in the placebo group. Filgotinib, another selective JAK1 inhibitor, had notable superiority over the placebo in the randomized, double-blind, phase 2 trial EQUATOR for PsA. That study revealed that a considerably higher proportion of patients who received filgotinib achieved the primary endpoint of ACR20 response at week 16. Deucravacitinib is a small molecule that selectively and non-competitively inhibits TYK2. By binding to the regulatory domain of TYK2, it modulates the immune inflammatory response to IL-12, IL-23, and type I IFNs [125][80]. Brepocitinib is an orally administered drug with dual inhibitory activity toward TYK2 and JAK1. New findings from a phase 2 randomized controlled trial indicated that both 30 mg and 60 mg doses of brepocitinib administered once daily were more effective in alleviating PsA signs and symptoms than the placebo.5. Summary
The progress in the elucidation of the impacts of inflammatory cytokines on PsA pathogenesis has enabled exploration of the novel molecular mechanisms that can be precisely targeted by therapeutic approaches. The activation of immunoinflammatory pathways in PsA involves multiple cell types and pro-inflammatory cytokines, such as TNF-α, IL-17, and IL-23. Providing timely and effective treatment is of the utmost importance in preventing the spread of joint damage and improving the overall well-being of individuals diagnosed with PsA. Despite the advancements in understanding PsA pathogenesis and the development of new drugs that target specific cytokines, managing PsA continues to pose challenges for rheumatologists. The pathogenesis and current therapeutic targets of PsA are shown in Figure 1. The ongoing studies investigating PsA pathogenesis offer the potential to reveal more pertinent therapeutic targets and broaden the array of available treatment options. Furthermore, there is a burgeoning interest in utilizing new biotechnology tools and combination therapies aimed at targeting multiple pathogenic pathways to achieve more effective disease control. Although there is little reliable clinical evidence of new drugs surpassing previous b/tsDMARDs in efficacy, significant advancements achieved in recent years in this field offer a promising outlook for the future.Figure 1.
Pathogenesis of PsA and therapeutic targets.
References
- Ritchlin, C.T.; Colbert, R.A.; Gladman, D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017, 376, 957–970.
- Schett, G.; Rahman, P.; Ritchlin, C.; McInnes, I.B.; Elewaut, D.; Scher, J.U. Psoriatic arthritis from a mechanistic perspective. Nat. Rev. Rheumatol. 2022, 18, 311–325.
- López-Medina, C.; Molto, A.; Sieper, J.; Duruöz, T.; Kiltz, U.; Elzorkany, B.; Hajjaj-Hassouni, N.; Burgos-Vargas, R.; Maldonado-Cocco, J.; Ziade, N.; et al. Prevalence and distribution of peripheral musculoskeletal manifestations in spondyloarthritis including psoriatic arthritis: Results of the worldwide, cross-sectional ASAS-PerSpA study. RMD Open 2021, 7, e001450.
- Qi, F.; Tan, Y.; Yao, A.; Yang, X.; He, Y. Psoriasis to Psoriatic Arthritis: The Application of Proteomics Technologies. Front. Med. 2021, 8, 681172.
- Veale, D.J.; Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 2018, 391, 2273–2284.
- Kane, D.; Stafford, L.; Bresnihan, B.; FitzGerald, O. A prospective, clinical and radiological study of early psoriatic arthritis: An early synovitis clinic experience. Rheumatology 2003, 42, 1460–1468.
- Haroon, M.; Gallagher, P.; FitzGerald, O. Diagnostic delay of more than 6 months contributes to poor radiographic and functional outcome in psoriatic arthritis. Ann. Rheum. Dis. 2015, 74, 1045–1050.
- Queiro, R.; Morante, I.; Cabezas, I.; Acasuso, B. HLA-B27 and psoriatic disease: A modern view of an old relationship. Rheumatology 2016, 55, 221–229.
- Muto, M.; Date, Y.; Ichimiya, M.; Moriwaki, Y.; Mori, K.; Kamikawaji, N.; Kimura, A.; Sasazuki, T.; Asagami, C. Significance of antibodies to streptococcal M protein in psoriatic arthritis and their association with HLA-A*0207. Tissue Antigens. 1996, 48, 645–650.
- Thorarensen, S.M.; Lu, N.; Ogdie, A.; Gelfand, J.M.; Choi, H.K.; Love, T.J. Physical trauma recorded in primary care is associated with the onset of psoriatic arthritis among patients with psoriasis. Ann. Rheum. Dis. 2017, 76, 521–525.
- Scher, J.U.; Ubeda, C.; Artacho, A.; Attur, M.; Isaac, S.; Reddy, S.M.; Marmon, S.; Neimann, A.; Brusca, S.; Patel, T.; et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015, 67, 128–139.
- Boix-Amorós, A.; Badri, M.H.; Manasson, J.; Blank, R.B.; Haberman, R.H.; Neimann, A.L.; Girija, P.V.; Jimenez Hernandez, A.; Heguy, A.; Koralov, S.B.; et al. Alterations in the cutaneous microbiome of patients with psoriasis and psoriatic arthritis reveal similarities between non-lesional and lesional skin. Ann. Rheum. Dis. 2022, 82, 507–514.
- Jadon, D.R.; Stober, C.; Pennington, S.R.; FitzGerald, O. Applying precision medicine to unmet clinical needs in psoriatic disease. Nat. Rev. Rheumatol. 2020, 16, 609–627.
- McGonagle, D. Enthesitis: An autoinflammatory lesion linking nail and joint involvement in psoriatic disease. J. Eur. Acad. Dermatol. Venereol. 2009, 23 (Suppl. S1), 9–13.
- Azuaga, A.B.; Ramírez, J.; Cañete, J.D. Psoriatic Arthritis: Pathogenesis and Targeted Therapies. Int. J. Mol. Sci. 2023, 24, 4901.
- Fitzgerald, O.; Winchester, R. Psoriatic arthritis: From pathogenesis to therapy. Arthritis Res. Ther. 2009, 11, 214.
- Candia, L.; Marquez, J.; Hernandez, C.; Zea, A.H.; Espinoza, L.R. Toll-like receptor-2 expression is upregulated in antigen-presenting cells from patients with psoriatic arthritis: A pathogenic role for innate immunity? J. Rheumatol. 2007, 34, 374–379.
- Jongbloed, S.L.; Lebre, M.C.; Fraser, A.R.; Gracie, J.A.; Sturrock, R.D.; Tak, P.P.; McInnes, I.B. Enumeration and phenotypical analysis of distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R15.
- Lew, W.; Bowcock, A.M.; Krueger, J.G. Psoriasis vulgaris: Cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. 2004, 25, 295–305.
- van Mens, L.J.J.; van de Sande, M.G.H.; Menegatti, S.; Chen, S.; Blijdorp, I.C.J.; de Jong, H.M.; Fluri, I.A.; Latuhihin, T.E.; van Kuijk, A.W.R.; Rogge, L.; et al. Brief Report: Interleukin-17 Blockade With Secukinumab in Peripheral Spondyloarthritis Impacts Synovial Immunopathology Without Compromising Systemic Immune Responses. Arthritis Rheumatol. 2018, 70, 1994–2002.
- Nigrovic, P.A.; Lee, D.M. Mast cells in inflammatory arthritis. Arthritis Res. Ther. 2004, 7, 1.
- Noordenbos, T.; Yeremenko, N.; Gofita, I.; van de Sande, M.; Tak, P.P.; Caňete, J.D.; Baeten, D. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis Rheum. 2012, 64, 99–109.
- Chen, S.; Noordenbos, T.; Blijdorp, I.; van Mens, L.; Ambarus, C.A.; Vogels, E.; Te Velde, A.; Alsina, M.; Cañete, J.D.; Yeremenko, N.; et al. Histologic evidence that mast cells contribute to local tissue inflammation in peripheral spondyloarthritis by regulating interleukin-17A content. Rheumatology 2019, 58, 617–627.
- Leijten, E.F.; van Kempen, T.S.; Boes, M.; Michels-van Amelsfort, J.M.; Hijnen, D.; Hartgring, S.A.; van Roon, J.A.; Wenink, M.H.; Radstake, T.R. Brief report: Enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol. 2015, 67, 2673–2678.
- Soare, A.; Weber, S.; Maul, L.; Rauber, S.; Gheorghiu, A.M.; Luber, M.; Houssni, I.; Kleyer, A.; von Pickardt, G.; Gado, M.; et al. Cutting Edge: Homeostasis of Innate Lymphoid Cells Is Imbalanced in Psoriatic Arthritis. J. Immunol. 2018, 200, 1249–1254.
- Sakkas, L.I.; Bogdanos, D.P. Are psoriasis and psoriatic arthritis the same disease? The IL-23/IL-17 axis data. Autoimmun. Rev. 2017, 16, 10–15.
- Qu, N.; Xu, M.; Mizoguchi, I.; Furusawa, J.; Kaneko, K.; Watanabe, K.; Mizuguchi, J.; Itoh, M.; Kawakami, Y.; Yoshimoto, T. Pivotal roles of T-helper 17-related cytokines, IL-17, IL-22, and IL-23, in inflammatory diseases. Clin. Dev. Immunol. 2013, 2013, 968549.
- Lories, R.J.; McInnes, I.B. Primed for inflammation: Enthesis-resident T cells. Nat. Med. 2012, 18, 1018–1019.
- Veale, D.J.; Ritchlin, C.; FitzGerald, O. Immunopathology of psoriasis and psoriatic arthritis. Ann. Rheum. Dis. 2005, 64 (Suppl. S2), ii26–ii29.
- Diani, M.; Altomare, G.; Reali, E. T cell responses in psoriasis and psoriatic arthritis. Autoimmun. Rev. 2015, 14, 286–292.
- Kirkham, B.W.; Kavanaugh, A.; Reich, K. Interleukin-17A: A unique pathway in immune-mediated diseases: Psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology 2014, 141, 133–142.
- Saddala, M.S.; Huang, H. Identification of novel inhibitors for TNFα, TNFR1 and TNFα-TNFR1 complex using pharmacophore-based approaches. J. Transl. Med. 2019, 17, 215.
- Zamri, F.; de Vries, T.J. Use of TNF Inhibitors in Rheumatoid Arthritis and Implications for the Periodontal Status: For the Benefit of Both? Front. Immunol. 2020, 11, 591365.
- de Vries, T.J.; El Bakkali, I.; Kamradt, T.; Schett, G.; Jansen, I.D.C.; D’Amelio, P. What Are the Peripheral Blood Determinants for Increased Osteoclast Formation in the Various Inflammatory Diseases Associated With Bone Loss? Front. Immunol. 2019, 10, 505.
- Kaaij, M.H.; van Tok, M.N.; Blijdorp, I.C.; Ambarus, C.A.; Stock, M.; Pots, D.; Knaup, V.L.; Armaka, M.; Christodoulou-Vafeiadou, E.; van Melsen, T.K.; et al. Transmembrane TNF drives osteoproliferative joint inflammation reminiscent of human spondyloarthritis. J. Exp. Med. 2020, 217, e20200288.
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390.
- Łukasik, Z.; Gracey, E.; Venken, K.; Ritchlin, C.; Elewaut, D. Crossing the boundaries: IL-23 and its role in linking inflammation of the skin, gut and joints. Rheumatology 2021, 60, iv16–iv27.
- Sherlock, J.P.; Cua, D.J. Interleukin-23 in perspective. Rheumatology 2021, 60, iv1–iv3.
- Huynh, D.; Kavanaugh, A. Psoriatic arthritis: Current therapy and future approaches. Rheumatology 2015, 54, 20–28.
- Maeda, S.; Hayami, Y.; Naniwa, T.; Ueda, R. The Th17/IL-23 Axis and Natural Immunity in Psoriatic Arthritis. Int. J. Rheumatol. 2012, 2012, 539683.
- Tam, H.K.J.; Robinson, P.C.; Nash, P. Inhibiting IL-17A and IL-17F in Rheumatic Disease: Therapeutics Help to Elucidate Disease Mechanisms. Curr. Rheumatol. Rep. 2022, 24, 310–320.
- Iznardo, H.; Puig, L. Dual inhibition of IL-17A and IL-17F in psoriatic disease. Ther. Adv. Chronic. Dis. 2021, 12, 20406223211037846.
- Millar, N.L.; Akbar, M.; Campbell, A.L.; Reilly, J.H.; Kerr, S.C.; McLean, M.; Frleta-Gilchrist, M.; Fazzi, U.G.; Leach, W.J.; Rooney, B.P.; et al. IL-17A mediates inflammatory and tissue remodelling events in early human tendinopathy. Sci. Rep. 2016, 6, 27149.
- Kavanaugh, A.; Mease, P.J.; Gomez-Reino, J.J.; Adebajo, A.O.; Wollenhaupt, J.; Gladman, D.D.; Lespessailles, E.; Hall, S.; Hochfeld, M.; Hu, C.; et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann. Rheum. Dis. 2014, 73, 1020–1026.
- McInnes, I.B.; Anderson, J.K.; Magrey, M.; Merola, J.F.; Liu, Y.; Kishimoto, M.; Jeka, S.; Pacheco-Tena, C.; Wang, X.; Chen, L.; et al. Trial of Upadacitinib and Adalimumab for Psoriatic Arthritis. N. Engl. J. Med. 2021, 384, 1227–1239.
- Miyagawa, I.; Nakayamada, S.; Ueno, M.; Miyazaki, Y.; Iwata, S.; Kubo, S.; Sonomoto, K.; Anan, J.; Ohkubo, N.; Inoue, Y.; et al. Impact of serum interleukin-22 as a biomarker for the differential use of molecular targeted drugs in psoriatic arthritis: A retrospective study. Arthritis Res. Ther. 2022, 24, 86.
- Min, H.K.; Won, J.-Y.; Kim, B.-M.; Lee, K.-A.; Lee, S.-J.; Lee, S.-H.; Kim, H.-R.; Kim, K.-W. Interleukin (IL)-25 suppresses IL-22-induced osteoclastogenesis in rheumatoid arthritis via STAT3 and p38 MAPK/IκBα pathway. Arthritis Res. Ther. 2020, 22, 222.
- Holt, V.E.; Luo, Y.; Colbert, R.A. IL-22 Effects on Osteoblast Mineralization and Metabolic Profile. J. Immunol. 2020, 204, 73–77.
- Kwon, O.C.; Park, M.C.; Kim, Y.G. Interleukin-32 as a biomarker in rheumatic diseases: A narrative review. Front. Immunol. 2023, 14, 1140373.
- Al-Shobaili, H.A.; Farhan, J.; Zafar, U.; Rasheed, Z. Functional role of human interleukin-32 and nuclear transcription factor-kB in patients with psoriasis and psoriatic arthritis. Int. J. Health Sci. 2018, 12, 29–34.
- Dong, Y.; Zhong, J.; Dong, L. IL-33 in Rheumatic Diseases. Front. Med. 2021, 8, 739489.
- Abdelaal, N.H.; Elhefnawy, N.G.; Abdulmonem, S.R.; Sayed, S.; Saleh, N.A.; Saleh, M.A. Evaluation of the expression of the stromal cell-derived factor-1 alpha (CXCL 12) in psoriatic patients after treatment with Methotrexate. J. Cosmet. Dermatol. 2020, 19, 253–258.
- Yager, N.; Cole, S.; Lledo Lara, A.; Maroof, A.; Penkava, F.; Knight, J.C.; Bowness, P.; Al-Mossawi, H. Ex vivo mass cytometry analysis reveals a profound myeloid proinflammatory signature in psoriatic arthritis synovial fluid. Ann. Rheum. Dis. 2021, 80, 1559–1567.
- Abji, F.; Pollock, R.A.; Liang, K.; Chandran, V.; Gladman, D.D. Th17 gene expression in psoriatic arthritis synovial fluid and peripheral blood compared to osteoarthritis and cutaneous psoriasis. Clin. Exp. Rheumatol. 2018, 36, 486–489.
- Sundanum, S.; Orr, C.; Veale, D. Targeted Therapies in Psoriatic Arthritis-An Update. Int. J. Mol. Sci. 2023, 24, 6384.
- Mantravadi, S.; Ogdie, A.; Kraft, W.K. Tumor necrosis factor inhibitors in psoriatic arthritis. Expert. Rev. Clin. Pharmacol. 2017, 10, 899–910.
- Wendling, D.; Joshi, A.; Reilly, P.; Jalundhwala, Y.J.; Mittal, M.; Bao, Y. Comparing the risk of developing uveitis in patients initiating anti-tumor necrosis factor therapy for ankylosing spondylitis: An analysis of a large US claims database. Curr. Med. Res. Opin. 2014, 30, 2515–2521.
- Levin, A.D.; Wildenberg, M.E.; van den Brink, G.R. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. J. Crohns Colitis 2016, 10, 989–997.
- Mease, P.J.; Gladman, D.D.; Collier, D.H.; Ritchlin, C.T.; Helliwell, P.S.; Liu, L.; Kricorian, G.; Chung, J.B. Etanercept and Methotrexate as Monotherapy or in Combination for Psoriatic Arthritis: Primary Results From a Randomized, Controlled Phase III Trial. Arthritis Rheumatol. 2019, 71, 1112–1124.
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; van der Heijde, D.; Landewé, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1137–1146.
- Nash, P.; Mease, P.J.; McInnes, I.B.; Rahman, P.; Ritchlin, C.T.; Blanco, R.; Dokoupilova, E.; Andersson, M.; Kajekar, R.; Mpofu, S.; et al. Efficacy and safety of secukinumab administration by autoinjector in patients with psoriatic arthritis: Results from a randomized, placebo-controlled trial (FUTURE 3). Arthritis Res. Ther. 2018, 20, 47.
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann. Rheum. Dis. 2017, 76, 79–87.
- Nash, P.; Kirkham, B.; Okada, M.; Rahman, P.; Combe, B.; Burmester, G.R.; Adams, D.H.; Kerr, L.; Lee, C.; Shuler, C.L.; et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: Results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet 2017, 389, 2317–2327.
- Ritchlin, C.T.; Kavanaugh, A.; Merola, J.F.; Schett, G.; Scher, J.U.; Warren, R.B.; Gottlieb, A.B.; Assudani, D.; Bedford-Rice, K.; Coarse, J.; et al. Bimekizumab in patients with active psoriatic arthritis: Results from a 48-week, randomised, double-blind, placebo-controlled, dose-ranging phase 2b trial. Lancet 2020, 395, 427–440.
- Mease, P.J.; Asahina, A.; Gladman, D.D.; Tanaka, Y.; Tillett, W.; Ink, B.; Assudani, D.; de la Loge, C.; Coarse, J.; Eells, J.; et al. Effect of bimekizumab on symptoms and impact of disease in patients with psoriatic arthritis over 3 years: Results from BE ACTIVE. Rheumatology 2023, 62, 617–628.
- Mease, P.J.; Helliwell, P.S.; Hjuler, K.F.; Raymond, K.; McInnes, I. Brodalumab in psoriatic arthritis: Results from the randomised phase III AMVISION-1 and AMVISION-2 trials. Ann. Rheum. Dis. 2021, 80, 185–193.
- Mease, P.J.; McInnes, I.B.; Tam, L.S.; Eaton, K.; Peterson, S.; Schubert, A.; Chakravarty, S.D.; Parackal, A.; Karyekar, C.S.; Nair, S.; et al. Comparative effectiveness of guselkumab in psoriatic arthritis: Results from systematic literature review and network meta-analysis. Rheumatology 2021, 60, 2109–2121.
- Fragoulis, G.E.; Siebert, S. The role of IL-23 and the use of IL-23 inhibitors in psoriatic arthritis. Musculoskeletal. Care 2022, 20 (Suppl. S1), S12–S21.
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 2013, 382, 780–789.
- Deodhar, A.; Helliwell, P.S.; Boehncke, W.-H.; Kollmeier, A.P.; Hsia, E.C.; Subramanian, R.A.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B.; et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2020, 395, 1115–1125.
- Mease, P.J.; Rahman, P.; Gottlieb, A.B.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B.; Zhuang, Y.; et al. Guselkumab in biologic-naive patients with active psoriatic arthritis (DISCOVER-2): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2020, 395, 1126–1136.
- Laura, C.C.; Laure, G.; Elke, T.; Paul, B.; Marlies, N.; Chetan, S.K.; May, S.; Wim, N.; Georg, S.; Iain, B.M. Efficacy and safety of guselkumab in patients with active psoriatic arthritis who are inadequate responders to tumour necrosis factor inhibitors: Results through one year of a phase IIIb, randomised, controlled study (COSMOS). Ann. Rheum. Dis. 2022, 81, 359.
- McInnes, I.B.; Rahman, P.; Gottlieb, A.B.; Hsia, E.C.; Kollmeier, A.P.; Xu, X.L.; Jiang, Y.; Sheng, S.; Shawi, M.; Chakravarty, S.D.; et al. Long-Term Efficacy and Safety of Guselkumab, a Monoclonal Antibody Specific to the p19 Subunit of Interleukin-23, Through Two Years: Results From a Phase III, Randomized, Double-Blind, Placebo-Controlled Study Conducted in Biologic-Naive Patients With Active Psoriatic Arthritis. Arthritis Rheumatol. 2022, 74, 475–485.
- Lars Erik, K.; Mauro, K.; Kim, P.; Leslie, M.; Douglas, W.; Wenjing, L.; Zailong, W.; Ahmed, M.S.; Ann, E.; Lisa, B.; et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 1 trial. Ann. Rheum. Dis. 2022, 81, 225.
- Andrew, Ö.; Van den Bosch, F.; Kim, P.; Cecilia, A.; Ricardo, B.; Jacob, A.; Gabriela, A.; Wenjing, L.; Zailong, W.; Ahmed, M.S.; et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 2 trial. Ann. Rheum. Dis. 2022, 81, 351.
- Campanaro, F.; Batticciotto, A.; Zaffaroni, A.; Cappelli, A.; Donadini, M.P.; Squizzato, A. JAK inhibitors and psoriatic arthritis: A systematic review and meta-analysis. Autoimmun. Rev. 2021, 20, 102902.
- Strand, V.; de Vlam, K.; Covarrubias-Cobos, J.A.; Mease, P.J.; Gladman, D.D.; Graham, D.; Wang, C.; Cappelleri, J.C.; Hendrikx, T.; Hsu, M.A. Tofacitinib or adalimumab versus placebo: Patient-reported outcomes from OPAL Broaden-a phase III study of active psoriatic arthritis in patients with an inadequate response to conventional synthetic disease-modifying antirheumatic drugs. RMD Open 2019, 5, e000806.
- Strand, V.; de Vlam, K.; Covarrubias-Cobos, J.A.; Mease, P.J.; Gladman, D.D.; Chen, L.; Kudlacz, E.; Wu, J.; Cappelleri, J.C.; Hendrikx, T.; et al. Effect of tofacitinib on patient-reported outcomes in patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors in the phase III, randomised controlled trial: OPAL Beyond. RMD Open 2019, 5, e000808.
- Nash, P.; Coates, L.C.; Kivitz, A.J.; Mease, P.J.; Gladman, D.D.; Covarrubias-Cobos, J.A.; FitzGerald, O.; Fleishaker, D.; Wang, C.; Wu, J.; et al. Safety and Efficacy of Tofacitinib in Patients with Active Psoriatic Arthritis: Interim Analysis of OPAL Balance, an Open-Label, Long-Term Extension Study. Rheumatol. Ther. 2020, 7, 553–580.
- Caso, F.; Costa, L.; Triggianese, P.; Maione, F.; Bertolini, N.; Vastarella, M.; Chimenti, M.S.; Tasso, M. Recent developments for new investigational JAK inhibitors in psoriatic arthritis. Expert. Opin. Investig. Drugs 2023, 32, 361–371.
More