Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alfredo Parra-Lucares and Version 2 by Sirius Huang.
Atrial fibrillation (AF) is a prevalent cardiac condition predominantly affecting older adults, characterized by irregular heartbeat rhythm. The condition often leads to significant disability and increased mortality rates.
- atrial fibrillation
- electrophysiology
1. Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia worldwide, with a global prevalence of around 1%, reaching nearly 10% in individuals over 80 years old [1]. It is characterized by a chaotic atrial rhythm derived from irregular electrical activity in different ectopic trigger sites, such as the pulmonary veins (PV) and the left atrium (LA) [2]. It is a significant cause of disability and death, especially in the elderly population, due to the development of complications, such as strokes, frequent hospitalizations, and bleeding associated with anticoagulant therapy [3]. This population is highly relevant, given that the primary risk factor for the onset of AF is age [4].
The therapeutic approach in AF has been a subject of debate for decades regarding the management of heart rate versus heart rhythm [5]. Initially, it was established that there was no difference between one therapeutic approach and another, leading to a preference for rate control over rhythm control due to the low rate of effectiveness and the proarrhythmic complications associated with antiarrhythmic drugs (AAD) [6]. However, over time, evidence began to demonstrate that AF progresses through different stages of the disease via electrical and structural remodeling phenomena that intensify with the passage of years [7]. Recent studies have highlighted the advantages of early conversion to sinus rhythm in patients with this arrhythmia [8][9][10][8,9,10].
Based on the above, it can be concluded that the restoration of sinus rhythm may reverse a series of electrophysiological and molecular mechanisms that underlie the natural progression of the disease, through which a patient transitions from paroxysmal AF episodes to the final stage of permanent AF [11].
2. The Hallmarks of Atrial Fibrillation
The clinical classification of AF is principally based on whether the episode terminates spontaneously or not, and the duration of the episode, progressing from paroxysmal, persistent, long persistent to permanent AF [1]. Recently, new classification systems for AF have been proposed [12][21]. Nonetheless, their pathophysiological correlate is still uncertain. The classical clinical categories have proven distinct behaviors that may reflect the underlying molecular mechanisms [13][22], although they are still not fully understood. This section will describe the pathophysiological continuum that explains the origin, maintenance, progression, and stabilization of AF, with the key aspects that have been described for each of the clinical stages, which will be referred to as “hallmarks”. Researchers also propose an “at risk” stage, analogous to what exists for heart failure (HF) [14][23] that is based on the genetic susceptibility and non-AF remodeling that may lead in some people to the development of this disease. It must be noted that AF has several clinical presentations that do not always follow this order (Figure 1).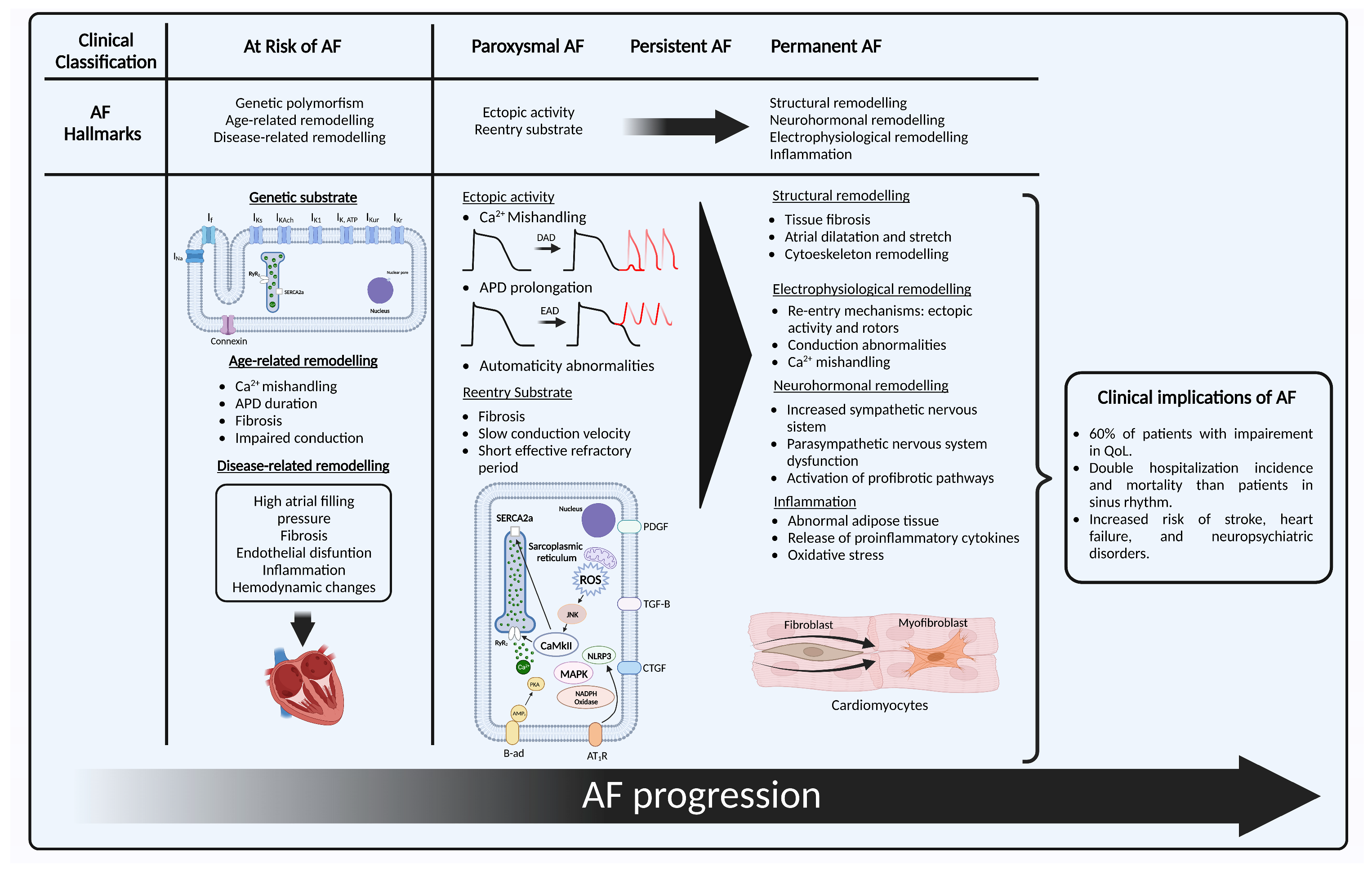
Figure 1. Representation of AF hallmarks with their clinical correlate. First, “at risk of AF” with its three hallmarks: (1) genetic substrate shows a cardiomyocyte with the main structures in which genetic variants for ion channel and non-ion channel genes that have shown association with the development of AF: IK1 current (KCNJ2), IKs (KCNE1, KCNE2, KCNE3, KCNE4, KCNE5, KCND3, KCNQ1), IKAch (KCNJ5), IKur (KCNA5), IKr (KCNH2), IKATP (ABCC9), If (HCN4), INa (SCN1B, SCN2B, SCN3B, SCN5A, SCN10A), connexin 40 (GJA5), connexin 43 (GJA1), and nucleoporin 155 (NUP155); (2) age-related remodeling; (3) disease-related remodeling; next to it the clinical stages of AF, with the initial hallmarks of paroxysmal AF: (1) ectopic activity secondary to DAD, EAD or automaticity abnormalities; (2) re-entry and some of its intracellular mechanisms of perpetuation; and the hallmarks of persistent and permanent AF that include structural, electrophysiological, and neurohormonal remodeling, and inflammation. Created with BioRender.com (accessed on 28 May 2023). AF: atrial fibrillation; DAD: delayed afterdepolarization; EAD: early afterdepolarization; APD: action potential duration; SERCA2a: sarcoplasmic reticulum Ca2+-adenosine triphosphate; RyR2: ryanodine receptors; CaMKII: Ca2+/calmodulin-dependent protein kinase II; PDGF: platelet-derived growth factor; TGF-β: transforming growth factor β; MAPK: mitogen-activated protein kinase; AMP: adenosine-monophosphate; β-ad: β-adrenergic receptor; AT1-R: angiotensin receptor 1; NLRP3: NACHT, LRR, and PYD domain containing protein 3 inflammasome; ROS: radical oxygen species.
2.1. At Risk for Atrial Fibrillation
2.1.1. Genetic Substrate
To develop AF, the well-known ectopic activity needs a histological and electrophysiological substrate. Several genetic polymorphisms and mutations have been identified that encode genes associated with ion channel function, calcium handling, transcription factors, and cardiac development and function that have been described elsewhere [15][12]. Nonetheless, for most people, these associations do not explain the entirety of the disease and only place them as a group that is at risk of developing AF [16][19]. Familial AF adds up to only 10–15% of AF cases [15][12], and generally present an earlier onset, in people that may lack traditional risk factors, which has also been called lone-AF. These monogenic paradigms have helped to gain understanding of the mechanisms that generate AF in an otherwise healthy tissue.2.1.2. Age-Related Remodeling
Age is one of the most well-known risk factors for developing AF, with a lifetime risk of 40% and 50% of patients with AF aged 75 and older [17][24]. Age-related cardiomyocyte loss produces fibrosis [18][25], and the automaticity in the sinoatrial node involves hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, whose expression ha shown an age-dependent increase in animal models, which may have a role in atrial ectopy [11]. Other electrophysiological and conduction changes that lead to Ca2+ mishandling, prolongation in the action potential duration (APD), effective refractory period (ERP) depolarization, and connexin downregulation also promote arrhythmogenesis [19][26]. In addition, older patients have a higher incidence of cardiovascular comorbidities like hypertension, HF, and valvular heart disease, and their atrial tissue has been exposed for a longer period to different stressors and remodeling processes [18][25].2.1.3. Disease-Related Remodeling
Environmental factors play a key role in the development of AF in most patients. It is estimated that hypertension, in particular high systolic blood pressure, is responsible for 14% of all cases of AF [20][27]. Obesity provides a proinflammatory, metabolic, and comorbid context that leads these patients to be at high risk of developing cardiovascular disease (CVD) [21][28]. Insulin resistance is linked to endothelial dysfunction that results from a mismatch between mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways, which increases endothelial cell death and inflammation [22][18], and in diabetes, hyperglycaemia leads to advanced glycation end products that lead to fibrosis and hypertrophy. Other factors like lifestyle and diet have shown harmful, but also potentially protective effects in susceptibility to AF [22][18]. Regarding cardiovascular comorbidities, HF is associated with increased atrial filling pressure that lead to atrial dilation and wall stretch. This histologically translates to a “tear and scar” mechanism that generates fibrosis [23][29]. A similar situation occurs with valvular heart diseases. Current classification includes subclinical AF defined as an individual without symptoms attributable to AF, in whom AF is not previously diagnosed and presents an atrial high-rate episode detected by an insertable cardiac or wearable monitor [1]. It is still not clear in which patients the use of devices for primary prevention might be beneficial, but many predictors and risk factors have been identified that include but are not limited to the aforementioned [24][30]. Guidelines recommend an opportunistic screening in patients with 65 years old or more [1]. Nonetheless, considering the rapidly growing wearable technology and artificial intelligence AF models, researchers believe that defining a group of patients at risk of AF, which has a clear pathophysiological basis, may prove useful for early detection strategies of AF.2.2. Paroxysmal Atrial Fibrillation
During normal atrial action potential, the cardiac voltage-dependent Na+ channel produces the depolarizing current (INa) that triggers the activation of L-type Ca2+ channels that is responsible for the calcium-induced calcium release from the sarcoplasmic reticulum (SR). Ca2+ is released by ryanodine receptors (RyR2), its uptake into the SR is mediated by the SR Ca2+-adenosine triphosphate (SERCA2a), and inside the SR it is bound to calsequestrin. SERCA2a function is limited by the inhibitory subunit phospholamban. Meanwhile, delayed-rectifier K+ currents (IKr, IKs, IKur) and the transient-outward K+ current (Ito) control the repolarization and determine the action potential duration (APD) [25][31]. The hallmarks in paroxysmal AF are the focal ectopic activity and a re-entry substrate prone to abnormal conduction, that are major determinants in AF onset and perpetuation [7]. PVs are the main source of ectopic activity in AF [26][32]. Their structure has branching fibres with limited lateral coupling and abrupt changes in their orientation. This provides an adequate anatomic substrate for spontaneous activity and re-entry. In patients with AF, PVs have shown a shorter effective refractory period (ERP), lower voltage, shorter muscle sleeves, slower conduction, and more complex signals [27][33]. These characteristics make them prone to develop delayed afterdepolarizations (DAD) or early afterdepolarizations (EAD) [11]. A longer APD allows L-type Ca2+ channels to recover from inactivation, which causes EAD. This may be due to loss of repolarizing K+ current, or a persistent or late Na+ current, such as in long-QT syndrome type 3 [28][34]. DADs are originated mainly from the abnormal release of Ca2+ from the SR during diastole, which is then exchanged for extracellular Na+ by the Na+-Ca2+ exchanger type 1 (NCX1) that leads to cell depolarization when it reaches a threshold [13][22]. Automatism abnormalities may also be a causing or perpetuating factor, since HCN channels that control automatism in the sinoatrial node have shown an increased expression in patients with AF. Nonetheless, their role in this disease is still not fully understood [11]. Regarding the second hallmark, the determinants for conduction are the structural integrity, cell-to-cell coupling, and the rapid phase of Na+-current. Alterations in these factors may come from fibrosis and fibroblasts differentiation into myofibroblasts that may promote ectopic activity; mutations of repolarizing K+ channels that produce a gain of function, some nucleoporin mutations (Nup155); and Na+ channel loss of function that can reduce the ERP and lead to a substrate favorable for re-entry [11]. The development of these two hallmarks is independent, and in some patients one of them may be dominant. Most of them respond well to pulmonary vein isolation (PVI), but some may experience recurrence [29][35]. When AF is established, it generates electrical and structural changes that promote its own maintenance, progression, and stabilization [30][31][36,37].2.3. Persistent Atrial Fibrillation
Persistent AF (PeAF) is defined as AF that is continuously sustained beyond 7 days [1]. The progression to this stage has been associated with cardiovascular events, hospitalizations, and death [32][38]. It has structural and electrophysiological differences by means of which AF can sustain itself and progress [33][39]. These are classified into four hallmarks: Structural remodeling, electrophysiological remodeling, neurohormonal remodeling, and inflammation.2.3.1. Structural Remodeling
Fibrosis plays a central role in the pathogenesis of AF. Atrial fibrous-tissue content, and therefore left atrial wall thickness, is increased in patients with PeAF and atrial scarring, and correlates with clinical outcomes [34][40]. It is classified in reparative and interstitial fibrosis. The first occurs due to the replacement of lost cardiomyocytes, while the second occurs in response to cardiac inflammation or pressure overload, and is sub-classified into reactive fibrosis, which indicates the deposition of extracellular matrix without cell replacement, and infiltrative fibrosis as seen in amyloidosis [22][35][16,18]. There are many profibrotic pathways that are upregulated in patients with AF [35][16]. Increased angiotensin II plays a key role in atrial remodeling by binding to angiotensin receptor 1 (AT1-R) that leads to the activation of the MAPK pathway, which regulates the expression of transforming growth factor β (TGF-β), connective tissue growth factor (CTGF) [36][41], plasminogen activator inhibitor (PAI-1), and matrix metalloproteinases, that are proinflammatory and profibrotic [35][16]. TGF-β promotes the synthesis of collagen fibres by cardiac fibroblasts and their differentiation into myofibroblasts. In addition, AT1-R is coupled with G protein that activates phospholipase C to mediate the increase in Ca2+ in the cytoplasm, which also promotes fibroblast proliferation and differentiation [37][42]. Experimental models have demonstrated that suppressing this pathway leads to a reduction in interstitial fibrosis by inhibiting both the mineralocorticoid receptor and the AT1-R [38][39][43,44]. This correlates with clinical trials, that have shown the efficacy of the inhibition of this axis for primary prevention of AF, especially in hypertension and heart failure populations, and in preventing AF recurrence after cardioversion and in patients with paroxysmal AF under medical therapy [40][41][42][43][45,46,47,48], which suggest a potential role for these therapeutic targets in AF secondary prevention. Nonetheless, this has not shown a reduction in mortality [44][49]. Another key component that suffers from AF remodeling is the sarcomeric cytoskeleton. This organelle supports mechanical contraction, signal transduction, and the transport of ubiquitinated proteins. AF generates mechanical stress and cytoskeletal protein damage that reach a failure in the protein quality control system [15][12]. Fibrosis leads to AF perpetuation in numerous ways. First, the excess of extracellular matrix creates a physical barrier for conduction, which induces re-entry. Second, the proliferation of fibroblasts and their differentiation into myofibroblasts lead to myofibroblast–cardiomyocyte interaction, that induce re-entry and spontaneous focal activity. Third, slowing conduction and APD reduction induce spontaneous depolarization. These alterations make the ideal substrate for AF.2.3.2. Electrophysiological Remodeling
The main components of this hallmark include a SR Ca2+ overload and Ca2+ spontaneous release. Adrenergic stimulation, AT-1R, and oxidative stress cause the activation of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) and protein kinase A (PKA), that phosphorylates RyR2 and phospholamban, increasing intracellular Ca2+. A reduced expression, hyperphosphorylation of the inhibitory interacting proteins phospholamban or atrial-specific sarcolipin lead to an increase in the activity of SERCA2a [11]. Meanwhile, a high atrial rate causes the accumulation of intracellular Ca2+, which activates the Ca2+-dependent calcineurin/nuclear factor of activated T cells (NFAT), that leads to a decreased Ca2+ current, which shortens the APD. Increased intracellular Ca2+ upregulates the expression of K+ channels, which also shorten the APD [45][50]. A reduced APD increases the likelihood of DAD that may lead to spontaneous atrial ectopic activity [25][31]. The intracellular spontaneous calcium release is favored by RyR2 dysfunction that increases its opening probability. This can be due to loss of RyR2-associated calmodulin, or of juntophilin-2. Its hyperphosphorylation by CaMKII at Ser2814, 2808 or 2030 by PKA promotes RyR2 dysfunction [11]. Uncertainty remains regarding the precise electrophysiological mechanism that sustains AF. Three main mechanisms have been described, and they are not mutually exclusive [46][51].-
Rotors: a re-entry mechanism that consists of a localized circular or spiral wavefront that rotates around an anatomical or functional obstacle, having heterogeneous conduction velocity and an unexcitable center that causes an irregular propagation of electrical activity.
-
Ectopic foci: abnormal regions within the atria that initiate electrical impulses spontaneously or in response to triggers.
-
Multiple wavelets: numerous re-entry circuits in the atria, that can interact with each other, merge, or divide.