Targeted cellular and immunotherapies have welcomed a new chapter in multi-modal cancer therapy. These agents harness our innate immune system and destroy malignant cells in a precise way as compared with “legacy” chemotherapeutic agents that largely rely on abolishing cell division. New therapies can augment the T-cell recognition of tumor antigens and effectively prevent tumor cells from their historically successful ability to evade immune recognition. These novel agents cause acute and chronic toxicities to a variety of organ systems (enteritis, pneumonitis, hypophysitis, and hepatitis), and this may masquerade as other chronic illnesses or paraneoplastic effects. As the perioperative footprint of cancer patients increases, it is essential that perioperative providers—anesthesiologists, surgeons, nurse anesthetists, and inpatient hospital medicine providers—be up to date on the physiologic mechanisms that underlie these new therapies as well as their acute and subacute toxicity profiles. Immunotherapy toxicity can significantly impact perioperative morbidity as well as influence perioperative management, such as prophylaxis for adrenal insufficiency, preoperative pulmonary assessment, and screening for thyroid dysfunction, among others.
- immunotherapy
- cellular therapy
- perioperative risk
- hypophysitis
1. Introduction
2. Immune-Checkpoint Inhibitors
Immune-checkpoint inhibitors have earned their keep as tremendously successful adjuncts to all manner of chemotherapeutic regimens. TheOur innate immune system relies on T-cell activation and proliferation to destroy foreign cells. T-cell activation requires both antigen–receptor coupling as well as co-stimulation by other immune effectors cells. This two-fold process underpins the versatility of theour immune system but is also the process by which cancer cells can evade detection. T-cell activation can be inhibited by two pathways: the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) pathway and the programmed cell death protein (PD-LA1) pathway. This interplay of T-cell activation and inhibition is referred to as an “immune checkpoint.” Cancer cells have evolved to thwart T-cell activation by taking advantage of the inhibition role of this immune checkpoint since CTLA-4 and PD-LA1 antigens are commonly found on the surfaces of tumor cells. Activation of these ligands by tumor cells allows them to evade detection. Immune-checkpoint inhibitors (ICIs), as a therapeutic target, prevent tumor cell evasion by acting as monoclonal antibodies against the CTLA-4 and PD-LA1 ligands. In preventing their ability to inactivate T cells, ICIs ensure that tumor cells are vulnerable to destruction (see Figure 1).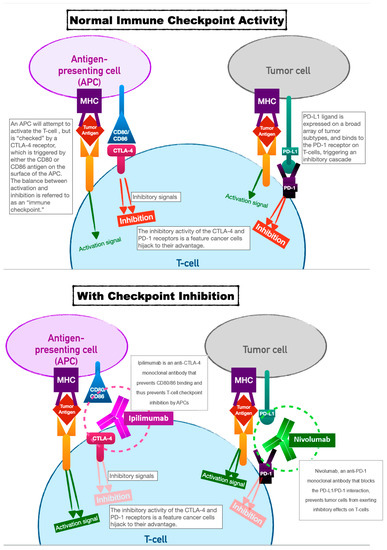
3. Chimeric Antigen-Receptor T Cells (CAR-T)
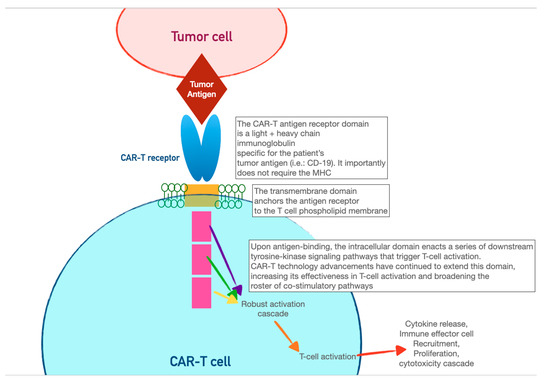
CAR-T cells harness the adaptive immunity offered by theour T-lymphocytes to target tumor-specific antigens. As the first foray into cellular therapeutics, they represent one of the most consequential discoveries in cancer therapy in the last two decades owing to their relative ease of administration and robust efficacy against highly fatal hematologic malignancies. As discussed in the previous section, the immune system relies on T-cell activation via (1) antigen presentation via an MHC complex and (2) co-stimulation via other immune effector cells. Cancer cells have historically evaded this process of T-cell activation by thwarting antigen recognition. Enter CAR-T cells: the genetically modified host T cells that force this process to occur without the ability of tumor-cell evasion. First, T cells are extracted from the patient (or an allogeneic donor) and modified via a retroviral vector to express a new antigen receptor and co-stimulatory domains (see Figure 2). The term “chimeric” refers to the fact that this engineered receptor both activates and co-stimulates the T cell [19][20][49,50]. The patient is then leuko-reduced to increase the relative proportion of CAR-T cells, and CAR-T cells are infused into the patient. The CAR-T cells will recognize antigens and activate and recruit immune effector cells to destroy tumor cells.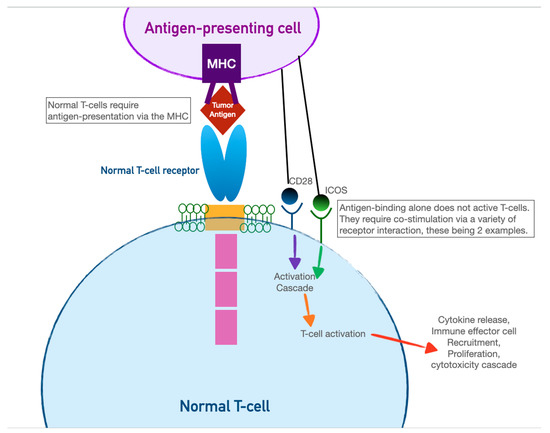
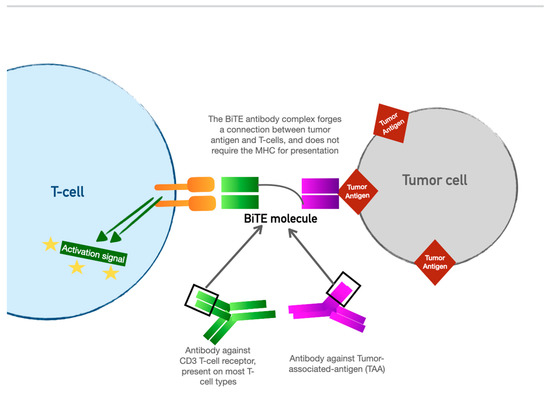
4. Bispecific T-Cell Engager (BiTE) Therapy
BiTE therapy is one of the newest innovations in targeted immunomodulatory chemotherapy. BiTE molecules are an antibody with two domains (hence “bispecific”): one that recognizes tumor-specific antigens and another that recognizes the universal CD-3 receptor on T cells (see Figure 3). The binding sites are essentially two monoclonal antibodies bound together. In essence, the BiTE “forces” a recognition and activation of the T cell, so the tumor cell does not have a chance to present its own antigen and possibly release inhibitory signal receptors. Specific tumor antigen targets of BiTE molecules include CD19 (broadly expressed on B-cell malignancies), B-cell maturation antigen (expressed highly on malignant cells involved in multiple myeloma), and CD33 (expressed in acute myeloid leukemia, myelodysplastic syndrome, and chronic myeloid leukemia). Under development are BiTE targets that include solid tumor antigens such as prostate-specific member antigen and delta-like protein 3, which is highly prevalent in small-cell lung cancer.