Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Xiaodong Li.
Clock proteins and their collaborating transcription factors often act as distal enhancers to regulate the rhythmic transcription of gene promoters. Those transcription factors need to interact with the mediator complex and general transcription factors near the transcription start site to finally control transcription. Pol II pausing, which is determined by Pol II recruitment, pause release, and premature transcription termination near the transcription start site, plays a critical role in influencing the final transcription output.
- circadian rhythms
- transcription
- pausing
1. Introduction
Molecular clockworks consisting of feedback loops of core clock genes drive cell-autonomous circadian oscillation in various species [1]. In mammals, the transcription factors (TFs) CLOCK and BMAL1 dimerize to activate the transcription of Per1/2 and Cry1/2, whose protein products are repressors that inhibit CLOCK/BMAL1 action through negative feedback [2]. While the post-translational regulation of clock proteins play critical roles in setting the clock pace [3[3][4],4], the prime mover of circadian oscillation is thought to be transcription [5]. High throughput technologies such as microarray [6], RNA-seq [7[7][8],8], and ChIP-seq [9] enable the detailed characterization of gene rhythms and the genomic binding of clock proteins, allowing for in depth analyses of circadian rhythm generation at the level of transcription.
The binding sites of clock proteins are located within open chromatin regions established by tissue-specific pioneer TFs (tsTFs), and thus are typically tissue-specific [10]. Chromatin is known to be a barrier to transcription, and DNA sequences are often not accessible to many TFs, with the exception of tsTFs that are sufficient to trigger enhancer competency within chromatin. Furthermore, tsTFs allow subsequent binding by other TFs, including clock proteins. Some tsTFs (e.g., HNF4a [11,12][11][12]) and ubiquitous TFs (u-TFs, e.g., RELA/p65 [13,14][13][14]) interact with and recruit clock proteins to their cis elements. CLOCK/BMAL1 can also facilitate the binding of some tsTFs, leading to the suggestion that CLOCK/BMAL1 acts like a pioneer-like TF [10,15][10][15]. Like many TFs [16], clock proteins recruit cofactors to modify histones and remodel nucleosomes to regulate transcription. Clock proteins and their cofactors form a complex with an M.W. over 1 MDa, and deficiencies in some cofactors alter clock dynamics [17]. For example, clock proteins in both Drosophila and mammals recruit the TIP60 complex to regulate clock oscillation [18,19,20][18][19][20]. To control Pol II transcription at the transcription start site (TSS), TFs require the mediator complex [21] to interact with general transcription factors (GTFs) that are present at gene promoter [22]. The mediator subunits interacting with the clock protein complex remain to be determined.
Traditional studies addressed how TFs and cofactors direct the mediator complex to assemble the pre-initiation complex (PIC) for transcription initiation and reinitiation at the TSS [23]. Distal enhancers bound by TFs and promoters are thought to be brought to proximity via chromatin looping, which is a process assisted by proteins such as cohesin and CTCF [24]. The traditional view of transcription, however, has difficulties in explaining new findings such as transcription bursting, which represents Pol II initiation and multiple rounds of reinitiation [25]. Imaging studies in single cells revealed that the transcription of many genes, including clock genes [26], is stochastic and of low frequency. Transcription often toggles between active and inactive states within a cell, and the active state is characterized by transcription bursting followed by a prolonged dormancy of the inactive state [27]. Bursting could be readily explained by the formation of a transcription hub: cluster and/or molecular condensate of TFs, cofactors, mediators, and Pol IIs that permit multiple rounds of Pol II initiation [28,29][28][29]. Recent studies revealed that TFs often contain intrinsically disordered low-complexity domains, whose interactions induce the formation of transcription clusters and even molecular condensates, the latter via “lipid-lipid phase separation” [28,29][28][29]. Transcription bursting also requires pause release [30], which refers to the process of P-TEFb-licensed Pol II elongation to overcome the +1 nucleosome barrier and transcribe into the gene body [31]. Initiated Pol II travels only a short distance; it then enters the state of pausing, wherein Pol II stays paused downstream of the TSS via the actions of pausing factors (DSIF and NELF) and the +1 nucleosome [31,32][31][32]. P-TEFb is a component of the super elongation complex (SEC) [33,34][33][34] that releases paused Pol II for elongation, permitting Pol II reinitiation to achieve transcription bursting. Initiated Pol II is also subject to premature transcription termination at the 5′ end of genes, which can decrease Pol II pausing [35,36][35][36].
2. Transcription Regulation Is the Main Driving Force for Gene Expression Rhythms
First demonstrated for Per in flies [39][37], core clock genes exhibit robust daily changes in their mRNA expression. Owing to rapid co-transcriptional splicing, the pre-mRNA level can be used as the surrogate for transcription activity. RNAse protection assays against Per pre-mRNA and mRNA showed that the Per mRNA rhythm in Drosophila is mainly driven at the level of transcription [40][38]. The post-transcriptional regulation of mRNA stability also contributes to the Per mRNA rhythm and is sufficient to confer rhythmic mRNA expression to other genes [41,42][39][40]. In mammals, core clock genes such as Per1/2 also exhibit robust daily changes in mRNA levels [43,44][41][42]. A pre-mRNA measurement implicated that rhythmic transcription is the driving force for the mRNA rhythms of core clock genes and many other genes [45,46][43][44]. Deep sequencing studies evaluated the contribution of transcription regulation to mRNA rhythm generation in a genome-wide manner. One study estimated that 22% of mRNA rhythms are driven by rhythmic transcription [9]. Later studies with a high sequencing depth and kinetic modeling increased the estimate to about 70–80%, whereas rhythmic degradation contributes to the mRNA rhythms of 30–35% genes [47,48][45][46]. Nuclear export, which is another post-transcriptional regulation step, contributes to rhythm generation for 10% of rhythmic transcriptomes [49][47]. Overall, rhythmic transcription is deemed as the main driving force for gene rhythms [5].3. Both the Intrinsic Tissue Clock and Extrinsic Cues Can Regulate Gene Expression Rhythms
Clock genes typically harbor multiple cis elements for clock proteins, which also have numerous other binding sites across the genome. Clock proteins thus also regulate many other genes. Clock genes and other genes are also influenced by extrinsic cues, which are often rhythmic in wildtype animals. Such cues include body temperature (Tb) [50][48], feeding [51][49], and communication signals from other tissues (including the autonomic nervous system) [52][50]. The extrinsic cues can engage TFs as well as post-transcriptional mechanisms to regulate gene rhythms, including those of clock genes. For example, daily changes in Tb drive rhythmic HSF1 expression to regulate gene transcription [53][51]. The Tb rhythm also drives Cirbp expression to post-transcriptionally regulate clock dynamics [54][52]. Besides the Tb rhythm, blood-borne cues also regulate clock dynamics; serum and plasma can activate multiple signaling pathways to impact clock genes [55,56,57][53][54][55]. For example, rhythmic cues in plasma activate SRF, which regulates the transcription of the clock gene mPer2 [57][55]. Certain blood-borne cues impacting clock dynamics are heat labile, implying that they are proteins [58][56]. Lipids can also serve as inter-tissue communicating cues. For example, phosphatidylcholine is synthesized by the liver and released into plasma to activate PPARα in muscles [59][57]. Overall, clock proteins and many other TFs exhibit daily changes in their actions. Like clock proteins, other TFs also have thousands of genomic binding sites in various tissues. Therefore, they potentially can regulate numerous genes besides clock genes. Gene expression rhythms are thus driven by both clock proteins and other TFs. How clock proteins and other TFs work together to control gene rhythms was the focus of recent studies in various peripheral mouse tissues.4. Clock Proteins Typically Collaborate with Other TFs to Regulate Transcription Rhythms
Clock proteins and other TFs often collaborate to regulate target genes [10]. The independent contribution of the clock to gene rhythms is rather limited [60,61][58][59]. In studies that reconstitute clock oscillation (RE) in specific tissues of Bmal1-deficient mice [60[58][59],61], it was shown that only 10% of the rhythmic transcriptome can be restored in the livers of liver-RE mice [60][58]. However, that is not to say that the liver clock regulates only 10% of the rhythmic transcriptome in wildtype mice. In fact, the disruption of the liver clock disturbs about 90% of the gene rhythms in mouse liver [62,63][60][61]. Overall, those results indicate that the majority of gene rhythms are regulated in a combinatorial manner by both the intrinsic clock and the TFs engaged by extrinsic cues. By comparing the liver gene rhythms in Bmal1 KO, liver-RE, and wildtype mice under ad libitum feeding versus nighttime restricted feeding, it was shown that the mRNA rhythms in the livers of the wildtype mice can be partitioned into four parts based on their modes of regulation [64][62]. Some rhythms can be driven by the intrinsic liver clock alone (13.7%); some can be driven by rhythmic feeding cues alone (17.5%); some require not only the intrinsic clock, but also rhythmic feeding cues (34.5%); while the rest (34.4%) require both the intrinsic clock and rhythmic cues from other tissues (and their clocks). Those results indicate that for the regulation of a majority of gene rhythms, there is a mandatory requirement for clock proteins to collaborate with other TFs. For example, feeding engages the TF CEBPB to coregulate BMAL1 target genes, and CEBPB deficiency disrupts the rhythms of some BMAL1 target genes that are also regulated by feeding [64][62].5. The Need to Study Pol II Pausing Regulation near the TSS
Clock proteins and other TFs occupying distinct enhancers of the same gene can collaborate through chromatin looping to regulate transcription. Techniques such as Hi-C and CHIA-PET revealed daily changes in the long-range interactions between distinct enhancers bound by clock proteins and other TFs, respectively, and between those enhancers and gene promoters [10,65][10][63]. The collaboration between clock proteins and other TFs can also occur at the same enhancers. Indeed, TFs often exhibit cooperative binding at the same enhancers to increase the affinities of the two factors to their respective motifs. However, cooperative binding does not necessarily lead to coactivation. For example, HNF4a and RELA/p65 can recruit CLOCK/BMAL1 for genomic binding [11[11][13],13], but can transrepress its transcription activation [12,14][12][14]. Such interactions between TFs at same and/or distinct enhancers pose serious challenges in elucidating how clock proteins contribute to the final transcription output at the TSS. Indeed, the genomic binding of CLOCK/BMAL1 at enhancers is often not sufficient to specify the rhythm phase and amplitude, and cannot confer rhythmicity to some target genes [37][64]. Another aspect of the complexity of transcription regulation is the lack of consensus on how TFs and their cofactors at distal enhancers regulate transcription near the TSS [66][65]. The textbook model of chromatin looping posits that a stable contact is formed between distal enhancers and promoters. A variation of this classical model is the “kiss-and-run” model of transient contact between distal enhancers and promoters. However, the nature of such long-range genomic interactions and its relevance to transcription have recently been questioned [66][65]. The alternative TAG (TF activity gradient) model [66][65] emphasizes contact-independent “communication by diffusion” of TFs and their cofactors between enhancers and promoters. However, given the diversity of interacting TFs and their multitudes of cofactors, it would be difficult, if not impossible, to dissect the specific contribution of an individual TF and/or cofactor to the transcription output. On the other hand, Pol II recruitment and Pol II pausing represent the final regulatory outcomes by a plethora of TFs and cofactors. Those regulatory steps are directly related to the final transcription output. Obtaining information about them is thus critical for understanding the logic of transcription regulation. Surprisingly, such information is lacking in circadian rhythm research (Figure 1).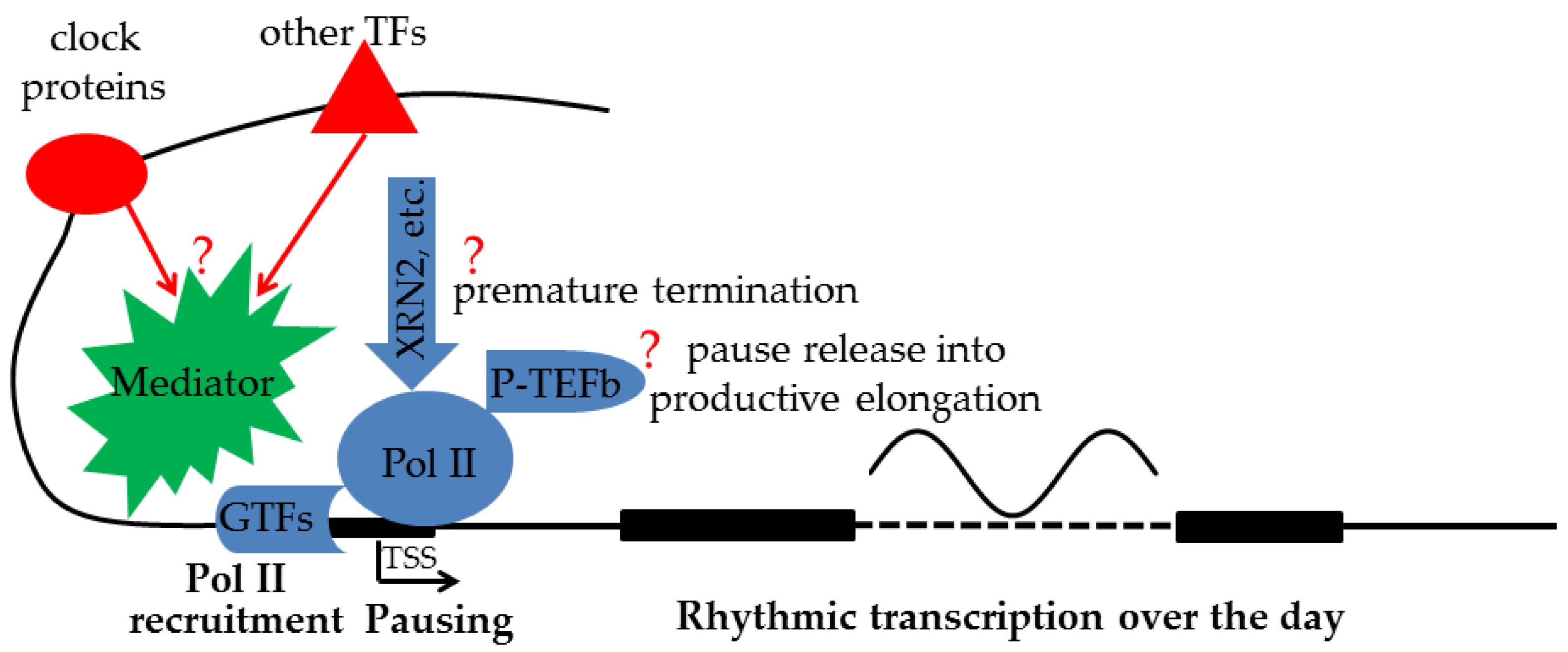
Figure 1. While clock proteins collaborate with other TFs at distal enhancers to regulate rhythmic transcription of target genes, exactly how final transcription output is determined by Pol II recruitment, premature termination, and pause release activities near the TSS is still an open question (?) and needs to be systematically characterized.
References
- Rosbash, M. The implications of multiple circadian clock origins. PLoS Biol. 2009, 7, e62.
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179.
- Zhou, M.; Kim, J.K.; Eng, G.W.; Forger, D.B.; Virshup, D.M. A period2 phosphoswitch regulates and temperature compensates circadian period. Mol. Cell 2015, 60, 77–88.
- Toh, K.L.; Jones, C.R.; He, Y.; Eide, E.J.; Hinz, W.A.; Virshup, D.M.; Ptacek, L.J.; Fu, Y.H. An hper2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 2001, 291, 1040–1043.
- Montenegro-Montero, A.; Larrondo, L.F. In the driver’s seat: The case for transcriptional regulation and coupling as relevant determinants of the circadian transcriptome and proteome in eukaryotes. J. Biol. Rhythm. 2016, 31, 37–47.
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320.
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224.
- Mure, L.S.; Le, H.D.; Benegiamo, G.; Chang, M.W.; Rios, L.; Jillani, N.; Ngotho, M.; Kariuki, T.; Dkhissi-Benyahya, O.; Cooper, H.M.; et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 2018, 359, eaao0318.
- Koike, N.; Yoo, S.H.; Huang, H.C.; Kumar, V.; Lee, C.; Kim, T.K.; Takahashi, J.S. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 2012, 338, 349–354.
- Beytebiere, J.R.; Trott, A.J.; Greenwell, B.J.; Osborne, C.A.; Vitet, H.; Spence, J.; Yoo, S.H.; Chen, Z.; Takahashi, J.S.; Ghaffari, N.; et al. Tissue-specific bmal1 cistromes reveal that rhythmic transcription is associated with rhythmic enhancer-enhancer interactions. Genes Dev. 2019, 33, 294–309.
- Qu, M.; Qu, H.; Jia, Z.; Kay, S.A. Hnf4a defines tissue-specific circadian rhythms by beaconing bmal1::Clock chromatin binding and shaping the rhythmic chromatin landscape. Nat. Commun. 2021, 12, 6350.
- Qu, M.; Duffy, T.; Hirota, T.; Kay, S.A. Nuclear receptor hnf4a transrepresses clock:Bmal1 and modulates tissue-specific circadian networks. Proc. Natl. Acad. Sci. USA 2018, 115, E12305–E12312.
- Hong, H.K.; Maury, E.; Ramsey, K.M.; Perelis, M.; Marcheva, B.; Omura, C.; Kobayashi, Y.; Guttridge, D.C.; Barish, G.D.; Bass, J. Requirement for nf-kappab in maintenance of molecular and behavioral circadian rhythms in mice. Genes Dev. 2018, 32, 1367–1379.
- Shen, Y.; Endale, M.; Wang, W.; Morris, A.R.; Francey, L.J.; Harold, R.L.; Hammers, D.W.; Huo, Z.; Partch, C.L.; Hogenesch, J.B.; et al. Nf-kappab modifies the mammalian circadian clock through interaction with the core clock protein bmal1. PLoS Genet. 2021, 17, e1009933.
- Menet, J.S.; Pescatore, S.; Rosbash, M. Clock: Bmal1 is a pioneer-like transcription factor. Genes Dev. 2014, 28, 8–13.
- Lemon, B.; Tjian, R. Orchestrated response: A symphony of transcription factors for gene control. Genes Dev. 2000, 14, 2551–2569.
- Aryal, R.P.; Kwak, P.B.; Tamayo, A.G.; Gebert, M.; Chiu, P.L.; Walz, T.; Weitz, C.J. Macromolecular assemblies of the mammalian circadian clock. Mol. Cell 2017, 67, 770–782.e776.
- Mahesh, G.; Rivas, G.B.S.; Caster, C.; Ost, E.B.; Amunugama, R.; Jones, R.; Allen, D.L.; Hardin, P.E. Proteomic analysis of drosophila clock complexes identifies rhythmic interactions with saga and tip60 complex component nipped-a. Sci. Rep. 2020, 10, 17951.
- Petkau, N.; Budak, H.; Zhou, X.; Oster, H.; Eichele, G. Acetylation of bmal1 by tip60 controls brd4-p-tefb recruitment to circadian promoters. Elife 2019, 8, e43235.
- Ju, D.; Zhang, W.; Yan, J.; Zhao, H.; Li, W.; Wang, J.; Liao, M.; Xu, Z.; Wang, Z.; Zhou, G.; et al. Chemical perturbations reveal that ruvbl2 regulates the circadian phase in mammals. Sci. Transl. Med. 2020, 12, eaba0769.
- Malik, S.; Roeder, R.G. The metazoan mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet. 2010, 11, 761–772.
- Thomas, M.C.; Chiang, C.M. The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178.
- Roeder, R.G. 50+ years of eukaryotic transcription: An expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol. 2019, 26, 783–791.
- Hsieh, T.S.; Cattoglio, C.; Slobodyanyuk, E.; Hansen, A.S.; Darzacq, X.; Tjian, R. Enhancer-promoter interactions and transcription are largely maintained upon acute loss of ctcf, cohesin, wapl or yy1. Nat. Genet. 2022, 54, 1919–1932.
- Richter, W.F.; Nayak, S.; Iwasa, J.; Taatjes, D.J. The mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2022, 23, 732–749.
- Suter, D.M.; Molina, N.; Gatfield, D.; Schneider, K.; Schibler, U.; Naef, F. Mammalian genes are transcribed with widely different bursting kinetics. Science 2011, 332, 472–474.
- Rodriguez, J.; Larson, D.R. Transcription in living cells: Molecular mechanisms of bursting. Annu. Rev. Biochem. 2020, 89, 189–212.
- Lim, B.; Levine, M.S. Enhancer-promoter communication: Hubs or loops? Curr. Opin. Genet. Dev. 2021, 67, 5–9.
- McSwiggen, D.T.; Mir, M.; Darzacq, X.; Tjian, R. Evaluating phase separation in live cells: Diagnosis, caveats, and functional consequences. Genes Dev. 2019, 33, 1619–1634.
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177.
- Chiu, A.C.; Suzuki, H.I.; Wu, X.; Mahat, D.B.; Kriz, A.J.; Sharp, P.A. Transcriptional pause sites delineate stable nucleosome-associated premature polyadenylation suppressed by u1 snrnp. Mol. Cell 2018, 69, 648–663.e647.
- Yamaguchi, Y.; Shibata, H.; Handa, H. Transcription elongation factors dsif and nelf: Promoter-proximal pausing and beyond. Biochim. Biophys. Acta 2013, 1829, 98–104.
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (sec) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547.
- Chen, Y.; Vos, S.M.; Dienemann, C.; Ninov, M.; Urlaub, H.; Cramer, P. Allosteric transcription stimulation by RNA polymerase II super elongation complex. Mol. Cell 2021, 81, 3386–3399.e3310.
- Price, D.H. Transient pausing by RNA polymerase II. Proc. Natl. Acad. Sci. USA 2018, 115, 4810–4812.
- Kamieniarz-Gdula, K.; Proudfoot, N.J. Transcriptional control by premature termination: A forgotten mechanism. Trends Genet. 2019, 35, 553–564.
- Hardin, P.E.; Hall, J.C.; Rosbash, M. Feedback of the drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 1990, 343, 536–540.
- Hardin, P.E.; Hall, J.C.; Rosbash, M. Circadian oscillations in period gene mrna levels are transcriptionally regulated. Proc. Natl. Acad. Sci. USA 1992, 89, 11711–11715.
- Frisch, B.; Hardin, P.E.; Hamblen-Coyle, M.J.; Rosbash, M.; Hall, J.C. A promoterless period gene mediates behavioral rhythmicity and cyclical per expression in a restricted subset of the drosophila nervous system. Neuron 1994, 12, 555–570.
- So, W.V.; Rosbash, M. Post-transcriptional regulation contributes to drosophila clock gene mrna cycling. EMBO J. 1997, 16, 7146–7155.
- Sun, Z.S.; Albrecht, U.; Zhuchenko, O.; Bailey, J.; Eichele, G.; Lee, C.C. Rigui, a putative mammalian ortholog of the drosophila period gene. Cell 1997, 90, 1003–1011.
- Shearman, L.P.; Zylka, M.J.; Weaver, D.R.; Kolakowski, L.F., Jr.; Reppert, S.M. Two period homologs: Circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron 1997, 19, 1261–1269.
- Preitner, N.; Damiola, F.; Lopez-Molina, L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The orphan nuclear receptor rev-erbalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260.
- Atger, F.; Gobet, C.; Marquis, J.; Martin, E.; Wang, J.; Weger, B.; Lefebvre, G.; Descombes, P.; Naef, F.; Gachon, F. Circadian and feeding rhythms differentially affect rhythmic mrna transcription and translation in mouse liver. Proc. Natl. Acad. Sci. USA 2015, 112, E6579–E6588.
- Luck, S.; Thurley, K.; Thaben, P.F.; Westermark, P.O. Rhythmic degradation explains and unifies circadian transcriptome and proteome data. Cell Rep. 2014, 9, 741–751.
- Wang, J.; Symul, L.; Yeung, J.; Gobet, C.; Sobel, J.; Luck, S.; Westermark, P.O.; Molina, N.; Naef, F. Circadian clock-dependent and -independent posttranscriptional regulation underlies temporal mrna accumulation in mouse liver. Proc. Natl. Acad. Sci. USA 2018, 115, E1916–E1925.
- Hurni, C.; Weger, B.D.; Gobet, C.; Naef, F. Comprehensive analysis of the circadian nuclear and cytoplasmic transcriptome in mouse liver. PLoS Genet. 2022, 18, e1009903.
- Buhr, E.D.; Yoo, S.H.; Takahashi, J.S. Temperature as a universal resetting cue for mammalian circadian oscillators. Science 2010, 330, 379–385.
- Damiola, F.; Le Minh, N.; Preitner, N.; Kornmann, B.; Fleury-Olela, F.; Schibler, U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000, 14, 2950–2961.
- Koronowski, K.B.; Sassone-Corsi, P. Communicating clocks shape circadian homeostasis. Science 2021, 371, eabd0951.
- Reinke, H.; Saini, C.; Fleury-Olela, F.; Dibner, C.; Benjamin, I.J.; Schibler, U. Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes Dev. 2008, 22, 331–345.
- Gotic, I.; Schibler, U. Posttranscriptional mechanisms controlling diurnal gene expression cycles by body temperature rhythms. RNA Biol. 2017, 14, 1294–1298.
- Balsalobre, A.; Marcacci, L.; Schibler, U. Multiple signaling pathways elicit circadian gene expression in cultured rat-1 fibroblasts. Curr. Biol. 2000, 10, 1291–1294.
- Balsalobre, A.; Damiola, F.; Schibler, U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 1998, 93, 929–937.
- Gerber, A.; Esnault, C.; Aubert, G.; Treisman, R.; Pralong, F.; Schibler, U. Blood-borne circadian signal stimulates daily oscillations in actin dynamics and srf activity. Cell 2013, 152, 492–503.
- Pagani, L.; Schmitt, K.; Meier, F.; Izakovic, J.; Roemer, K.; Viola, A.; Cajochen, C.; Wirz-Justice, A.; Brown, S.A.; Eckert, A. Serum factors in older individuals change cellular clock properties. Proc. Natl. Acad. Sci. USA 2011, 108, 7218–7223.
- Liu, S.; Brown, J.D.; Stanya, K.J.; Homan, E.; Leidl, M.; Inouye, K.; Bhargava, P.; Gangl, M.R.; Dai, L.; Hatano, B.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554.
- Koronowski, K.B.; Kinouchi, K.; Welz, P.S.; Smith, J.G.; Zinna, V.M.; Shi, J.; Samad, M.; Chen, S.; Magnan, C.N.; Kinchen, J.M.; et al. Defining the independence of the liver circadian clock. Cell 2019, 177, 1448–1462.e1414.
- Welz, P.S.; Zinna, V.M.; Symeonidi, A.; Koronowski, K.B.; Kinouchi, K.; Smith, J.G.; Guillen, I.M.; Castellanos, A.; Furrow, S.; Aragon, F.; et al. Bmal1-driven tissue clocks respond independently to light to maintain homeostasis. Cell 2019, 177, 1436–1447.e1412.
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.W.; DiTacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by rev-erb-alpha and rev-erb-beta. Nature 2012, 485, 123–127.
- Kornmann, B.; Schaad, O.; Bujard, H.; Takahashi, J.S.; Schibler, U. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biol. 2007, 5, e34.
- Greco, C.M.; Koronowski, K.B.; Smith, J.G.; Shi, J.; Kunderfranco, P.; Carriero, R.; Chen, S.; Samad, M.; Welz, P.S.; Zinna, V.M.; et al. Integration of feeding behavior by the liver circadian clock reveals network dependency of metabolic rhythms. Sci. Adv. 2021, 7, eabi7828.
- Kim, Y.H.; Marhon, S.A.; Zhang, Y.; Steger, D.J.; Won, K.J.; Lazar, M.A. Rev-erbalpha dynamically modulates chromatin looping to control circadian gene transcription. Science 2018, 359, 1274–1277.
- Beytebiere, J.R.; Greenwell, B.J.; Sahasrabudhe, A.; Menet, J.S. Clock-controlled rhythmic transcription: Is the clock enough and how does it work? Transcription 2019, 10, 212–221.
- Karr, J.P.; Ferrie, J.J.; Tjian, R.; Darzacq, X. The transcription factor activity gradient (tag) model: Contemplating a contact-independent mechanism for enhancer-promoter communication. Genes Dev. 2022, 36, 7–16.
- Zhu, J.; Li, C.; Gong, C.; Li, X. Regulation of pol II pausing is involved in daily gene transcription in the mouse liver. J. Biol. Rhythm. 2018, 33, 350–362.
- Yudkovsky, N.; Ranish, J.A.; Hahn, S. A transcription reinitiation intermediate that is stabilized by activator. Nature 2000, 408, 225–229.
- Hawley, D.K.; Roeder, R.G. Functional steps in transcription initiation and reinitiation from the major late promoter in a hela nuclear extract. J. Biol. Chem. 1987, 262, 3452–3461.
- Zawel, L.; Kumar, K.P.; Reinberg, D. Recycling of the general transcription factors during RNA polymerase II transcription. Genes Dev. 1995, 9, 1479–1490.
- Zhang, Z.; Boskovic, Z.; Hussain, M.M.; Hu, W.; Inouye, C.; Kim, H.J.; Abole, A.K.; Doud, M.K.; Lewis, T.A.; Koehler, A.N.; et al. Chemical perturbation of an intrinsically disordered region of tfiid distinguishes two modes of transcription initiation. Elife 2015, 4, e07777.
- Shao, W.; Zeitlinger, J. Paused RNA polymerase II inhibits new transcriptional initiation. Nat. Genet. 2017, 49, 1045–1051.
- Gressel, S.; Schwalb, B.; Cramer, P. The pause-initiation limit restricts transcription activation in human cells. Nat. Commun. 2019, 10, 3603.
- Tantale, K.; Mueller, F.; Kozulic-Pirher, A.; Lesne, A.; Victor, J.M.; Robert, M.C.; Capozi, S.; Chouaib, R.; Backer, V.; Mateos-Langerak, J.; et al. A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat. Commun. 2016, 7, 12248.
- Day, D.S.; Zhang, B.; Stevens, S.M.; Ferrari, F.; Larschan, E.N.; Park, P.J.; Pu, W.T. Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome Biol. 2016, 17, 120.
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. C-myc regulates transcriptional pause release. Cell 2010, 141, 432–445.
More