Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Shin Hyeong Sub and Version 2 by Lindsay Dong.
Long noncoding RNAs (lncRNAs) are molecules >200 bases in length without protein-coding functions implicated in signal transduction and gene expression regulation via interaction with proteins or RNAs, exhibiting various functions. The expression of lncRNAs has been detected in many cell types, including macrophages, a type of immune cell involved in acute and chronic inflammation, removal of dead or damaged cells, and tissue repair. Increasing evidence indicates that lncRNAs play essential roles in macrophage functions and disease development.
- atherosclerosis
- inflammation
- lncRNA
- macrophage
- sepsis
1. Introduction
Recent advances in molecular biology have revealed that although noncoding RNAs (ncRNAs) are not translated into proteins, they play various roles in cellular processes and disease pathogeneses. Long ncRNAs (lncRNAs) are >200 nucleotides in length and have been extensively researched in various fields of biology [1]. Nuclear lncRNAs have been implicated in regulating chromatin organization, gene transcription, RNA splicing, and epigenetic modifications [2][3][4][5][2,3,4,5]. Certain lncRNAs that exhibit structural features similar to those of mRNAs can be transported to the cytoplasm to modulate signaling pathways and post-transcriptional gene expression regulation by affecting mRNA stability and translation or sponging microRNAs (miRNAs) to block their function [6][7][8][9][10][6,7,8,9,10].
Macrophages exhibit immunoregulatory functions during acute and chronic inflammation, pathogenesis of various diseases, and cancer development. They are categorized into M1 and M2 functional groups. M1 macrophages (also known as “killer” or classically activated macrophages) phagocytose pathogens and foreign substances and promote inflammation, whereas M2 macrophages (also known as “repair” or alternatively activated macrophages) mediate tissue repair and inflammation resolution [11][12][11,12]. M1 macrophages metabolize arginine to nitric oxide and synthesize ATP via glycolysis. Furthermore, the mitochondrial citric acid cycle is shut down in these cells. Conversely, M2 macrophages metabolize arginine into ornithine or proline and mostly synthesize ATPs via the citric acid cycle [13]. The bacterial endotoxin lipopolysaccharide (LPS) and interferon (IFN)-γ induce the differentiation of undifferentiated (M0) macrophages into M1 macrophages, which produce a range of proinflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1, and IL-12. These cytokines and M1 macrophages have been associated with acute inflammation and tissue damage. Conversely, M2 differentiation, induced by IL-4 and IL-13, is characterized by the production of anti-inflammatory cytokines, such as IL-10 and transforming growth factor (TGF)-β. These cytokines and M2 macrophages help regulate immune responses and promote tissue repair [13][14][13,14]. Given that the activation and differentiation pathways and immunological roles of M1 and M2 macrophages differ, a detailed understanding of the factors that maintain or regulate the balance between them is important to effectively treat various diseases, such as autoimmune diseases, inflammatory bowel diseases, diabetes, obesity, rheumatoid arthritis (RA), and systemic sclerosis [13][15][13,15].
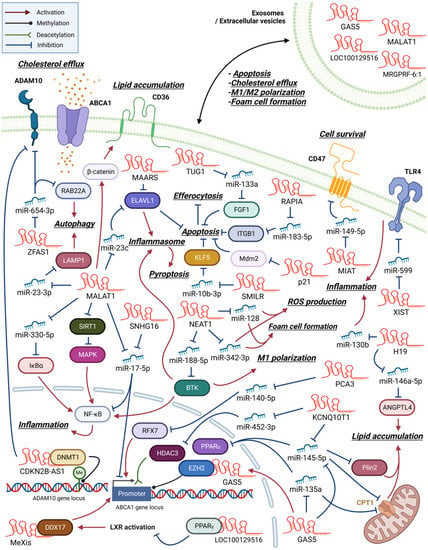
2. Atherosclerosis
Atherosclerosis is a chronic inflammatory disease characterized by the narrowing and hardening of arteries due to the accumulation of lipid-laden plaques on their inner walls. Macrophages engulf modified low-density lipoprotein (LDL) particles, such as oxidized LDL (oxLDL), and differentiate into foam cells, thereby aggravating chronic inflammatory conditions, stimulating plaque growth, and destabilizing plaques [16][17][18,19]. Atherosclerosis has been associated with various health problems, including coronary artery diseases (CADs) and stroke. M1 and M2 macrophages have been implicated in atherogenesis. M1 macrophages promote inflammation and plaque rupture by producing proinflammatory cytokines, chemokines, reactive oxygen species (ROS), and extracellular matrix-degrading enzymes. Conversely, M2 macrophages resolve inflammation by releasing anti-inflammatory cytokines, such as IL-10 [13][18][19][13,20,21].2.1. LncRNAs That Promote Inflammation and Foam Cell Formation
Many macrophage lncRNAs involved in atherosclerosis development are induced by oxLDL and promote foam cell formation through enhancing inflammatory changes (Figure 12). Previous studies found that lncRNA H19 expression was upregulated after oxLDL treatment in peripheral blood mononuclear cells of patients with CAD, plaque macrophages of an atherosclerotic mouse model, and RAW264.7 murine macrophage-like cells [20][21][22,23]. Transfecting H19-specific short hairpin RNA (shRNA) into RAW264.7 cells decreased oxLDL-induced lipid accumulation and proinflammatory mediator expression by regulating miR-130b activity [22][24].Figure 12. An overview of macrophage lncRNAs involved in atherosclerosis development. LncRNAs exert their functions through both direct and indirect mechanisms. Certain lncRNAs act as miRNA sponges, indirectly regulating protein expression by inhibiting miRNA function. In contrast, other lncRNAs directly interact with proteins to modulate their activity. Additionally, some lncRNAs can regulate gene expression through epigenetic modifications. Within the figures, arrows are employed to indicate the molecule responsible for activating or inhibiting its specific target.
NEAT1 (LINC00084, shortened from either nuclear paraspeckle assembly transcript 1 or nuclear enriched abundant transcript 1) is a nucleus-restricted lncRNA involved in the formation of paraspeckles, which are subnuclear structures implicated in antiviral responses [23][26]. NEAT1 expression is upregulated in oxLDL-treated THP-1 cells, a human monocytic leukemia cell line with macrophage-like properties. Furthermore, NEAT1 participates in the formation of paraspeckles and the development of subsequent proinflammatory responses by regulating p65 phosphorylation. Additionally, NEAT1 modulates lipid uptake by regulating the expression of a scavenger receptor, CD36 [24][27]. The treatment of RAW264.7 cells with oxLDL increases NEAT1 expression, which in turn stimulates proinflammatory cytokine and ROS production, subsequently promoting foam cell formation by sponging miR-128 [25][28].
In THP-1 cells, oxLDL treatment upregulated lncRNA urothelial cancer-associated 1 (UCA1) expression levels, which further exacerbated atherosclerotic events, such as CD36 expression, foam cell formation, and ROS generation via sponging miR-206 [26][32]. In atherosclerotic animal models, lncRNA dynamin 3 opposite strand (Dnm3os) expression was increased in atherosclerotic plaques. Dnm3os regulated macrophage proinflammatory activities via the miR-27b-3p/signaling lymphocytic activation molecule 7 (SLAMF7) axis [27][33]. SLAMF7 is a membrane protein whose expression is upregulated in macrophages during phagocytosis and macrophage differentiation in atherosclerotic plaques [28][34].
2.2. LncRNAs That Regulate Cholesterol Efflux and Foam Cell Formation
Numerous clinical and animal studies have demonstrated that defects in reverse cholesterol transport and cholesterol efflux are associated with an increased risk of cardiovascular diseases and atherosclerosis [16][17][18,19]. ATP-binding cassette subfamily A member 1 (ABCA1)-mediated cholesterol efflux reduces the formation of lipid-laden foam cells in atherosclerotic plaques. Because regulation of cholesterol efflux is crucial for the prevention of atherosclerosis, ABCA1 has been one of the primary targets in lncRNA research.
The lncRNA macrophage-expressed liver X receptor (LXR)-induced sequence (MeXis) is reportedly involved in LXR-dependent transcriptional activation of Abca1 by guiding the promoter binding of the transcription coactivator DEAD-box helicase 17 (DDX17). Furthermore, bone marrow cells from MeXis-deficient mice exhibited altered chromosome architecture at the Abca1 locus, impaired cholesterol efflux, and accelerated atherosclerosis development. Notably, the genes encoding ABCA1 and MeXis are located near one another to ensure tissue-selective activation of this regulatory circuit [29][35].
In contrast, the lncRNA growth arrest-specific 5 (GAS5) exerts inhibitory effects on ABCA1 function through its interaction with and stabilization of the enhancer of zeste homolog 2 (EZH2), a chromatin-repressive complex known to promote trimethylation of lysine 27 (H3K27) at the Abca1 promoter [30][38]. Notably, a significant elevation in GAS5 levels was detected in the serum of patients with coronary heart disease, exhibiting a correlation with heightened proinflammatory markers [31][39].