Some non-pathogenic viruses that do not cause serious illness in humans can efficiently target and kill cancer cells and may be considered candidates for cancer treatment with virotherapy. However, many cancer cells are protected from viruses. An important goal of personalized cancer treatment is to identify viruses that can kill a certain type of cancer cells. To this end, researchers investigate expression patterns of cell entry receptors, which viruses use to bind to and enter host cells.
- Oncolytic Virotherapy
- Cancer
- Oncolytic viruses
- Oncolytic measles virus
- Oncolytic Sendai virus
1. Introduction
Oncolytic viruses are promising new agents for cancer treatment. They can kill cancer cells directly through infection or indirectly through activation of the immune system [1][2][1,2]. For the most effective virotherapy, elimination of malignant cells with a combination of both direct and indirect destruction is desirable. Like all viruses, oncolytic viruses use specific receptors to bind to and enter host cells. This review describes the tendency of tumor cells to overexpress certain viral receptors, but it also shows that, to varying degrees, these receptors are also expressed in many normal cells. However, regardless of whether cells are normal or malignant, absence of receptors for a particular virus makes the cells resistant to this virus infection. So, for better identification of individual patients who are most likely to benefit from virotherapy, their tumor cells should be screened for the presence of virus receptors. For many oncolytic viruses, such receptors are well characterized. Thus, simple tests that evaluate protein or RNA levels in tumor tissue could provide information about expression levels of a virus receptor.
Receptor mediated virus entry into a cell is only the first step in viral infection. Next, the virus must break through the cellular antiviral defense system, which usually effectively protects normal cells from any virus infection. Key players in such protection are interferons (IFNs); they help cells detect the presence of a virus and, in response, restrict proliferation, slow down metabolic processes, and trigger apoptosis [3][4][3,4]. However, malignant cells frequently have dysfunctional IFN pathways. Such dysfunction helps them to evade the immune system and survive, thus promoting tumor growth. The same IFN defects that help cancer cells escape immune surveillance make them vulnerable to virus infection [5]. Nevertheless, not all malignant cells have dysfunctional IFN pathways. Some of them can produce and/or respond to IFN signals and protect themselves from a virus infection. So, theoretically, even if a cancer cell had receptors for a particular oncolytic virus it still could be resistant to infection by the virus.
Some viruses require cells to express processing enzymes that modify or cleave the viral proteins necessary for the formation of mature infectious virions. Thus, fusion protein in paramyxoviruses is synthesized as an inactive precursor and is activated through proteolytic cleavage by the cellular protease. Without such cleavage the virus is unable to sustain infection. For MV, this activating protease is furin [6] and for SeV it can be a number of serine proteases (TPSB2 [7][8][9][7,8,9], PRSS1 [10], PLG [11], F10 [12], and TMPRSS2 [13]). Some of these proteases are overexpressed in cancer cells [14][15][16][14,15,16]. In addition to those listed, the expression levels of other host genes influence vulnerability of cancer cells to a virus infection. FigureFigure 1 1 illustrates factors necessary for a cell to become vulnerable to paramyxovirus infection.

To predict if a patient is likely to respond to oncolytic virotherapy, testing for the presence of virus receptors in tumor tissue is not sufficient. Additional tests are also needed to reveal the presence of impaired IFN signaling in the patient’s cancer cells, and the expression of virus processing enzymes and yet to be identified other proteins that accommodate virus infection. Currently, such tests are commercially unavailable and need to be developed to optimize patient selection protocols for future clinical trials.
Oncolytic paramyxoviruses might become powerful anticancer agents [17][18][19][17,18,19]. FigureFigure 2 2 shows the life cycle of the viruses, which can trigger syncytium (a polykarion) formation that protects virions from host neutralizing antibodies during intratumor virus replication and spreading.
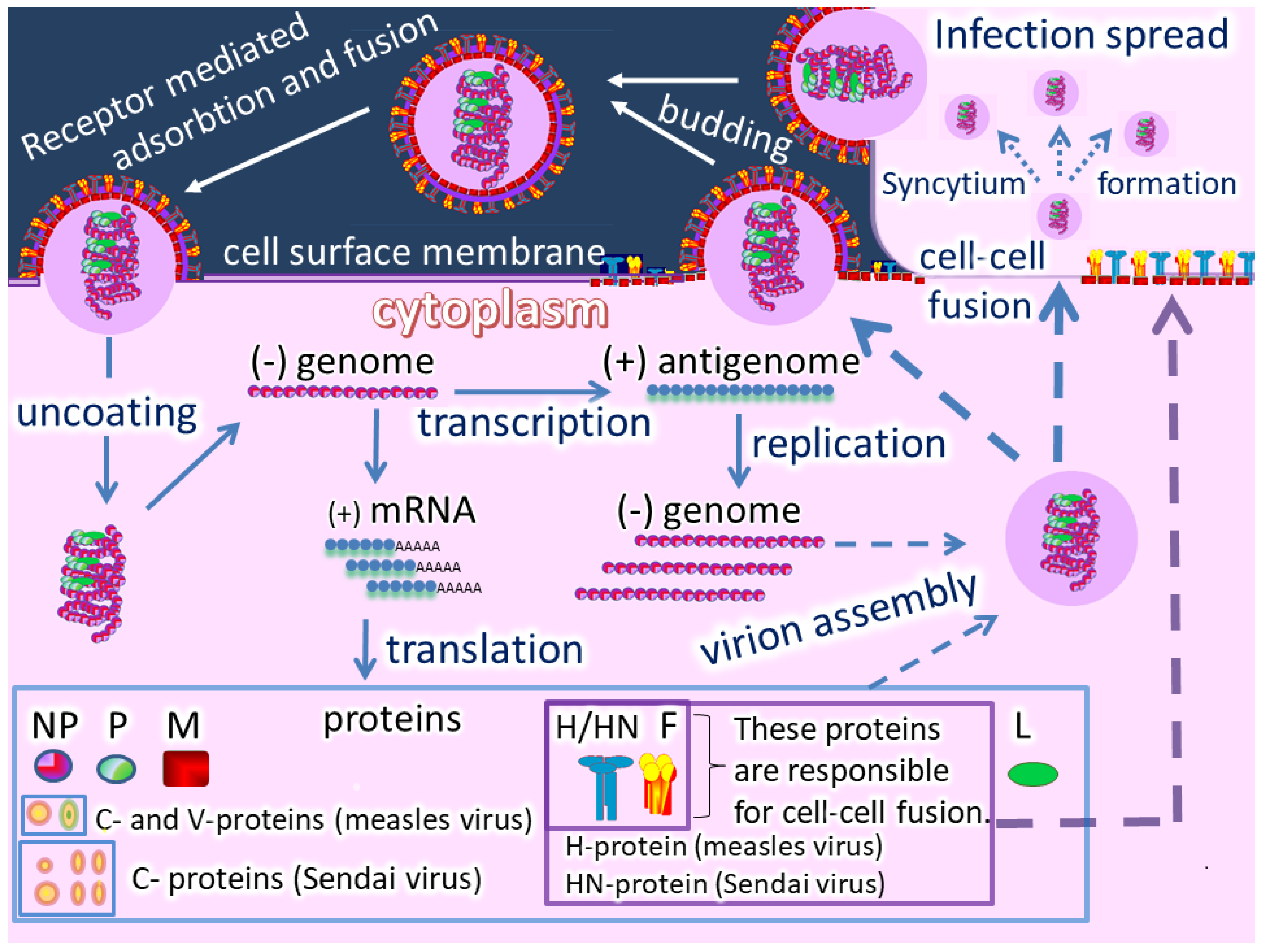
So, two related processes can occur: Efficient intratumor virus spread and the resulting mass death of malignant cells. In general, oncolytic paramyxoviruses stimulate strong innate and adaptive anticancer immune responses by generating multiple danger signals. They are potent inducers of IFN and other immuno-stimulating cytokines, and they efficiently induce anticancer activity of natural killer cells, dendritic cells, and cytotoxic T lymphocytes [18]. Finally, the viruses require proteolytic cleavage of their fusion proteins by cellular serine proteases, which are sometimes overexpressed in cancer cells [14][15][16][14,15,16], and could add an additional level of specificity to viral oncolytic activity. Moreover, the gene that encodes a fusion protein in the paramyxovirus genome can be replaced with a constructed fusion protein that could be processed by tumor-associated matrix metalloproteases.
2. Measles Virus as an Oncolytic Agent
MV (Box 1, Figure 3) causes a highly contagious disease transmitted by respiratory aerosols that can trigger severe immunosuppression and even immune amnesia.

Box 1. Measles Virus (MV).
|
Significant efforts by virologists in the second half of the 20th century were focused on finding a safe and effective vaccine against MV. In 1954, one MV isolated strain, when passaged in cell culture, gradually lost its pathogenicity, and became attenuated. From this attenuated variant of the virus, one of the first vaccine strain (Edmonston, denoted in the following text as MV-Edm) was obtained. Further passages of MV-Edm generated the more attenuated Schwarz and Moraten strains, which are still in use for vaccination against measles [23][24][24,25].
During the 20th century clinicians reported on isolated cases where measles disease relieved or caused remission of certain malignancies. Reports include descriptions of the regression of a number of hematological malignancies that took place after MV infection (reviewed in [25][26]). At the end of the century, virologists and oncologists started to investigate oncolytic properties of attenuated MV strains that are incapable of causing serious infection.
MV-Edm and its derivatives were the primary strains tested as oncolytic agents [26][27][27,28]. These strains can infect and kill a wide variety of cancer cells in vitro and in vivo. They are currently being investigated preclinically and in clinical trials for treatment of a large spectrum of malignancies, including ovarian, breast, head and neck cancers, as well as glioblastoma, multiple myeloma, mesothelioma, and T-cell lymphoma [26][27][27,28]. A virus with oncolytic properties can be delivered not only locally intratumorally, but also systemically, including by intraperitoneal and intravenous injection routes. In some trials patients survival compared favorably with that of historical controls and the side effects of the virotherapy were mainly mild [26][27][27,28].
3. Sendai virus as an oncolytic agent
Sendai virus (SeV) causes respiratory infections in mice and other rodents (Box 2). However, it is not associated with any human disease and can be a safe oncolytic agent. Safety of SeV for humans, including small children, has been confirmed experimentally. The virus in the form of nasal drops has been tested as a vaccine against human parainfluenza virus type 1, which causes respiratory symptoms in humans. Testing in adults and children demonstrated that experimental administration of wild type SeV triggered production of neutralizing antibodies towards human parainfluenza virus type 1 and was well tolerated [28][29] [29],[30]. SeV showed an excellent safety profile in a clinical trial [30][31].
Box 2. Sendai Virus (SeV)
|
Wild-type SeV can infect a large spectrum of malignant cells ex vivo [34][35]. A pilot study demonstrated that some canine (dog) cancers such as mastocytomas can be eradicated with the help of SeV injections [35][36]. However, the virus is infectious for laboratory rodents. Therefore, for studying SeV oncolytic properties in a rodent model, a set of viral constructs that are nonpathogenic for experimental mice was created. They were tested in animals bearing a variety of human xenograft tumors including sarcoma, melanoma, pancreas, colon, hepatocellular and prostate carcinomas. The SeV constructs promoted growth suppression or even complete tumor eradication of these malignancies [36][37][38][39][37][38][39][40], and elimination of established brain tumors [40][41].
In addition, UV- inactivated SeV virions have immune-stimulating properties: in animal model and they promote tumor regression of colon [41][42][42][43], bladder [43][44] and kidney [44][45] cancers. Other animal models these virions contribute to the eradication of human prostate cancer [45][46].
Oncolytic mechanism of Sendai virus action is discussed in detail in a number of publications [46][47][48][47][48][49].
So far, SeV has not been widely tested in clinical settings. An attempt to treat one case of human leukemia with a set of viruses, including SeV, was undertaken in 1964 at the Clinical Research Center of University Hospitals of Cleveland. Short-term remission in one patient affected by acute leukemia was observed after intravenous virus injection [49][50]. A few patients affected by various malignancies were treated with intradermally or intratumorally injected SeV in Moscow (Russia) in the 1990s. In a small fraction of patients treated with the virus, primary tumors and metastases disappeared, even when virotherapy was a monotherapy. These patients experienced a pronounced long-term remission that sometimes lasted more than 5-10 years [50][51].
4. Conclusions
The cell entry receptors of oncolytic paramyxoviruses are represented by different types of molecules. They are proteins for measles virus and sialylated proteins or glycans for Sendai virus.The molecules that serve as viral receptors for these viruses have extremely variable expression patterns in malignancies. A reliable predictive model for categorization of tumor cells according to their susceptibility to oncolytic virus infection requires many input parameters, which include but are not limited to expression patterns of viral receptors. Most likely, gene signatures of several immune-related genes such as IRGs and certain type II transmembrane serine proteases may serve as useful input parameters for predictive models. The creation and verification of a multi-parameter predictive model should have measurable therapeutic benefits for oncolytic virotherapy.