Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Natalia Todosenko.
Metabolic syndrome (MetS) is a precursor to the major health diseases associated with high mortality in industrialized countries: cardiovascular disease and diabetes. An important component of the pathogenesis of the metabolic syndrome is mitochondrial dysfunction. The mitochondrial AAA + protease Lon (Lonp1) has a broad spectrum of activities. In addition to its classical function (degradation of misfolded or damaged proteins), enzymatic activity (proteolysis, chaperone activity, mitochondrial DNA (mtDNA) binding) has been demonstrated.
- metabolic syndrome
- obesity
- mitochondrial dysfunction
- mtDNA
1. Introduction
Each cell contains many mitochondria, and each mitochondria contains several copies of mtDNA [11][1]. mtDNA is located in the mitochondrial matrix in close proximity to the electron transport chain, the major source of reactive oxygen species. Therefore, mtDNA is particularly susceptible to oxidation leading to mutations and the development of various pathologies [12][2]. Human mtDNA is 16,569 bp in size and contains 37 genes encoding 22 transfer RNAs, two ribosomal RNAs required for protein synthesis, and 13 messenger RNAs required for OXPHOS [13][3].
The double-stranded closed mtDNA molecule consists of a heavy outer ring (H) and a complementary light inner ring (L) that contain no introns [14][4]. At the same time, the D-loop (control region) is the most intense regulatory non-coding region of mtDNA, which contains the origin of replication of the H-strand. It is noted that the mitochondrial control region is subject to a high rate of mtDNA changes (especially in hypervariable regions) [13][3].
2. The Relationship between Quantitative mtDNA Levels (and Inflammation) in Obesity and Metabolic Syndrome
The mechanism underlying mtDNA CN alteration in metabolic disorders is directly related to excess energy substrates leading to insulin resistance mediated by an increase in inflammatory cytokine profile, resulting in mitochondrial fragmentation, oxidative stress (increased ROS production), accumulation of mtDNA damage, and cellular senescence [1][5]. Notably, there is evidence for a causal relationship between changes in mtDNA CN and inflammation in adipose tissue and oxidative stress. In adipose tissue, a decrease in mtDNA CN can lead to a deficiency in OXPHOS proteins and promote the formation of a pool of M1 macrophages that generate ATP through glycolysis and produce pro-inflammatory mediators, contributing to insulin resistance [15,16][6][7].
The tissue-specific indicator of the amount of mtDNA CN per cell is not a direct marker of mitochondrial function but is related to mitochondrial enzyme activity and adenosine triphosphate production [17][8]. This confirms the results of a study in which cells with reduced mtDNA CN had reduced expression of vital complex proteins, altered cell morphology, and lower respiratory enzyme activity [18][9].
Mitochondrial dysfunction has been shown to be associated with changes in the quantitative content of mtDNA and the degree of mtDNA damage [19][10] at MetS, insulin resistance, and obesity [20,21][11][12].
Studies in humans and animals have shown that loss of mtDNA content occurs in metabolic diseases [22][13]. The change in mtDNA CN in obesity has been described in studies that analyzed this parameter in adipose tissue [23][14] and peripheral blood [9][15].
Studies confirm a dynamic relationship between mtDNA CN and weight changes [24,25][16][17]. At the same time, a cross-sectional analysis of large cohorts showed a direct association between reduced mtDNA CN and obesity, diabetes, dyslipidemia, and hypertension [26][18].
A decrease in mtDNA CN in leukocytes, skeletal muscle cells, hepatocytes, and white adipocytes correlates with visceral obesity, body mass index, hyperlipidemia, cardiovascular disease, MetS, and mortality [5,27,28][19][20][21]. The results of studies with a limited sample of subjects diagnosed at MetS showed depletion of mtDNA CN in peripheral blood leukocyte cells [29,30][22][23].
In addition, lower levels of mtDNA CN correlated with MetS components in the general population [1,29,30][5][22][23].
In a recent study of a large cohort of patients with MetS and type 2 diabetes (type 2 DM), a strong association was found between low whole blood mtDNA CN levels and obesity, MetS, and type 2 DM. Moreover, mtDNA CN may predict the risk of developing MetS in patients with chronic kidney disease and in the European population as a whole [31][24].
Interestingly, a 10-year study in patients with MetS found an annual decrease of about five copies of mtDNA (in peripheral blood cells) [1][5], consistent with similar studies [6,32][25][26]. These studies draw a parallel between metabolic disorders and aging, as similar changes (decrease) in mtDNA copies per cell with age are observed in peripheral blood leukocytes [6][25], human pancreatic cells [33][27], fibroblasts, and myocytes.
In addition, higher triglyceride levels have been found to be associated with a decrease in mtDNA in peripheral blood mononuclear cells in patients with MetS [1][5], and similar associations have been shown for hyperglycemia and mtDNA [34][28].
A low baseline mtDNA CN was prospectively associated with a higher risk of developing type 2 DM [35][29]. It is noted that mtDNA CN level is a dynamic indicator and may change depending on endogenous/exogenous conditions. Thus, a higher mtDNA CN level was observed in type 2 diabetic patients with greater weight loss while taking the antidiabetic drug (metformin), which was associated with a decrease in glucose level, allowing the use of mtDNA CN for effective weight control in type 2 DM [36][30]. In addition, sex-specific variation in mtDNA CN was noted, as were the dynamics of this indicator before and after bariatric surgery [37][31]. In contrast, a decrease in mtDNA CN in peripheral blood was associated with an increase in mtDNA copy number in subcutaneous adipose tissue and TNF-a production in people with a high body mass index [23][14].
Thus, the above data suggest a direct link between mtDNA CN alterations and the development of metabolic disorders such as obesity, type 2 DM, and MetS and confirm the important role of mitochondrial dysfunction in the development of these diseases.
3. mtDNA Methylation
A close relationship has been established between mitochondria/mtDNA and nuclear DNA.
The mtDNA is essential for normal mitochondrial function, and many mitochondrial proteins are encoded by nuclear genes [38][32]. The interaction between mitochondria and the nuclear epigenome is bidirectional: mitochondria mediate epigenetic processes, and changes in the epigenome control mitochondrial function and influence stress response and lifespan [39,40][33][34].
Mitochondria provide for the formation of intermediate metabolites necessary for epigenetic modifications in the nucleus that control mitochondrial protein expression.
Interestingly, mtDNA CN also regulates nuclear gene expression through changes in nuclear CpG methylation, which leads to more severe pathology by regulating signaling pathways [41][35]. Epigenetic changes in nuclear DNA and low levels of mtDNA CN have been found to correlate with lower cancer survival [41][35].
This is of particular interest because the measurement of DNA methylation in peripheral blood cells can be used as a marker of less accessible tissues involved in obesity [42][36], and differences in peripheral blood cell DNA methylation profiles have been demonstrated in obesity [43][37].
The phenomenon of mtDNA methylation was discovered in the 1970s. CpG and non-CpG methylations were detected in mtDNA, and the presence of methyltransferases in mitochondria was also demonstrated. At the same time, in the human mitochondrial genome, the number of C sites (subject to methylation) of the L chain is more than twice the number of C sites of the H chain, with a small number of the C sites being dinucleotide CpG. Thus, methylation of mtDNA occurs mainly at the L chain and CpG sites. Moreover, 12 of 13 protein-coding genes use the L-chain as a template. L-chain methylation could affect mitochondrial function by controlling mtDNA gene expression [44][38].
The mechanism of methylation due to mtDNA changes suggests the involvement of histones. Thus, retrograde signaling from mitochondria to the nucleus regulates histone acetylation and alters gene expression in the nucleus through heterogeneous ribonucleoprotein A2 (hnRNAP2). At the same time, histone modifications correlate with mitochondrial content and are associated with chromatin activation (H4K16, H3L4me3, H3K36me2) [45][39]. Moreover, oxidative phosphorylation injury alters methylation processes due to modifications of the methionine cycle involved in the formation of S-adenosylmethionine, a methyl donor for histones and DNA methyltransferases [41][35].
Mutations and single nucleotide polymorphisms occur more frequently in the mitochondrial displacement loop (D-loop) than in other mtDNA regions [46][40]. This is mediated by the high sensitivity of the D-loop of mtDNA to ROS-dependent damage.
Functional regulation of mtDNA in metabolic disorders has been shown to be related to methylation marks in mtDNA (particularly in the bias loop, D-loop) [39][33].
Oxidative stress leads to increased expression of a key transcription factor that controls mitochondrial gene expression and mtDNA replication, mitochondrial transcription factor A (TFAM). TFAM initiates mitochondrial transcription by binding to the D-loop region and also regulates the process of mtDNA replication.
At the same time, D-loop methylation of mtDNA also affects mitochondrial gene expression and mtDNA replication [47][41]. High D-loop methylation disrupts TFAM, thereby reducing mitochondrial gene transcription and altering mitochondrial function [48,49][42][43]. A change in mtDNA copy number was found in response to an increase/decrease in the level of D-loop methylation [50,51][44][45]. Hypomethylation of the D-loop enhances gene expression [49][43] and leads to an increase in mtDNA copy number. Hypermethylation was associated with a decrease in mtDNA copy number [52][46].
According to the literature, mammalian mtDNA is enriched in N6-methyldeoxyadenosine (6 mA), presumably involving the methyltransferase METTL4, which causes suppression of mtDNA binding to TFAM. Hypoxia contributed to a 1300-fold increase in 6 mA labeling in mtDNA compared with total DNA. Furthermore, 6 mA mtDNA methylation promoted attenuation of mtDNA transcription, decreased mtDNA CN (HepG2 cells), and impaired mitochondrial activity [53][47].
A paper by Bordoni L. et al. [54][48] shows that mtDNA methylation patterns in DNA samples from buccal swabs are associated with body composition in overweight children, confirming previous studies on sex differences in mtDNA CN and obesity [5,37][19][31].
However, another paper by Bordoni L. et al. [55][49] found slight associations between mtDNA methylation levels and obesity, with no association between mtDNA methylation and mtDNA CN in VAT biopsies. At the same time, higher D-loop methylation was observed in severe obesity [55][49].
Studies show a direct association between changes in mtDNA methylation and the development of obesity [56][50] and cardiovascular disease [57][51].
High levels of mtDNA-D loop methylation in peripheral blood were found to be associated with low mtDNA-CN levels in insulin resistance and obesity [58][52].
Thus, mtDNA methylation may have a modulatory effect on the expression of important genes involved in the maintenance of mitochondrial and cellular function in the background of metabolic disorders.
4. mtDNA Mutations Accumulated during Development or in Postmitotic Tissues
The mitochondrial genome has a high mutation rate, about 100–1000 times higher than that of the nuclear genome. The mtDNA is not protected by histone proteins, so defective genes accumulate in it 10–20 times faster than in nuclear DNA [59][53].
Pathological mutations in mtDNA persist through mitochondrial fusion/fission processes and impaired mitophagy [60,61][54][55]. Accumulation of somatic mutations in mtDNA (heteroplasmy) leads to disruption of mitochondrial functions [62][56].
mtDNA mutations play an important role in the pathogenesis of MetS.
Mutations of tRNA genes are localized in areas important for tRNA stability, tRNA concentration in equilibrium in mitochondria, and aminoacylation activity, leading to high ROS production, development of oxidative stress, and apoptosis [59][53]. Mutations in the mitochondrial tRNA allele T4291C have been observed in the European population and have been associated with MetS and metabolic defects. In addition, mtDNA D-loop mutations, particularly T16189C, have been associated with insulin resistance and coronary heart disease [63,64][57][58]. Diabetes-associated mtDNA mutations: tRNALeu(UUR) A3243G heteroplasmy is involved in the pathogenesis of hereditary diabetes [65][59]. Studies on the role of the A3243G mutation have shown a direct link to mitochondrial dysfunction associated with inefficient aminoacetylation, impaired mRNA precursor processing, and basic posttranscriptional modification of tRNALeu(UUR) [66][60]. A study of the relationship of mitochondrial heteroplasmy in peripheral blood cells in a man with juvenile (as well as familial) diabetes, MetS, and a history of multiple sclerosis showed interesting results. Analysis of the mtDNA genome revealed a heteroplasmic mutation of mtDNA A8890G exclusively in the patient studied (but not in his relatives), whereas an association between the MetS and the single nucleotide polymorphism T16189C was found in all family members [67][61].
Thus, investigating the role of mtDNA polymorphisms in the pathogenesis of MetS and its components is a promising direction that will enable future work to prevent and predict the likelihood of developing severe pathologies and reduce the high risk of mortality (outcomes).
5. mtDNA Haplogroups
mtDNA variants are inherited maternally without recombination and can accumulate over time. A mitochondrial haplogroup groups together people who have the same accumulated mtDNA variants and may be geographically separated. In other words, haplogroups are ancient functional polymorphisms associated with specific mtDNA lineages [68][62]. Different haplogroups form different branches of the mitochondrial phylogenetic tree [13][3].
Studies have shown the association between mitochondrial haplogroups and the development of obesity [69,70,71][63][64][65] and MetS [72][66] in different parts of the world.
Studies have shown the association between Asian haplogroups (Japanese, Korean) and type 2 DM and MetS. Haplogroup N9a was associated with a low risk of type 2 DM and haplogroups D4/D5 and F with a high risk of type 2 DM in a cohort of Japanese and Koreans [73][67]. The Japanese also showed an association between haplogroup M8a (8684C˃T) (polymorphisms of single mitochondrial nucleotides) and high risk for type 2 DM and between B4c (3497C˃T, 1119T˃C) and obesity [74][68]. On the contrary, in the Chinese population, haplogroup N9a (N9a1, N9a10a) is predisposed to the development of type 2 DM [75][69]. Haplogroups M9 [76][70] and M8a [77][71] were markers of increased risk for type 2 DM in Han Chinese. A study in the Taiwanese population showed a protective role against the development of type 2 DM haplogroup D4 and a predisposing role in terms of B4a1a, E2b1 [78,79][72][73]. An association between a predisposition to type 2 DM and the presence of D4 H haplogroups was found in the Chinese Uighur population [80][74].
At the same time, the study by Chalkia D. et al. [72][66] showed the association between mitochondrial haplogroups and the risk of developing MetS.
Therefore, further studies on the relationship between mitochondrial haplogroups and the development of MetS will provide more clarity on the molecular features of the pathogenesis of the disease and allow the identification of specific targets to prevent the development of severe complications.
6. mtDNA Released from Cells as Alarmins and Inflammation Inducers
According to the “endosymbiont theory” proposed by Lynn Margulis, it is the evolutionary transformation of an autonomous endosymbiont bacterium into a specialized cell organelle, the mitochondrion, that results in mitochondrial products being potentially recognized by the immune system as “foreign” molecules [81][75]. The pro-inflammatory function of mtDNA was first demonstrated in 2004 [82][76]. Moreover, oxidation is an essential component of the mtDNA-associated inflammatory response [83,84][77][78]. When mtDNA binds to TFAM (mitochondrial transcription factor), the nucleotide is very stable; otherwise, mtDNA is in a fragile and easily degraded state. The study showed that cell-free mtDNA and TFAM-associated mtDNA play important roles in the inflammatory response [85][79].
Mitochondria-released mtDNA (which, like bacterial DNA, contains unmethylated CpG motifs) is a key factor in the activation of innate immunity mediated by pathogen-associated pattern recognition receptor (PRR) (PAMP) signaling cascades. These include the following signaling pathways: cGAS (cyclic GMP-AMP synthase pathway), Toll-like receptors (TLR9), NOD-like NLR receptors, the inflammasome NRLP3 (protein-3 with NACHT, LRR, and PYD domains) [86][80], and retinoic acid-inducible gene 1 (RIG-1)-like receptors (RLRs) and C-type lectin receptors (CLRs) [12,87,88,89][2][81][82][83].
Recurrent sterile inflammation (chronic production of pro-inflammatory cytokines), mediated in part by the release of mtDNA, is a classic example of a pathophysiological condition that occurs in patients with metabolic diseases [90][84]. It is interesting to note that in the elderly (over 90 years of age), plasma mtDNA levels gradually increase with age and correlate positively with plasma levels of TNF-a, IL-6, IL-1ra, RANTES [91][85]. Given the important pathogenic role of (high) circulating cytokines (TNF-a, IL-6, IL-1) and their association with poor disease prognosis [92][86], it is important to understand the mechanisms of association between mtDNA and the development of an inflammatory response in obesity and MetS.
Uncovering the role of mtDNA in MetS-associated inflammatory processes will therefore allow the identification of potential targets for early diagnosis and the prevention of severe disease sequelae.
7. Signaling Pathways Activated by mtDNA in Obesity and Metabolic Syndrome
7.1. cGAS-STING Signaling Pathway
Interferon gene stimulator (STING) is an inflammatory endoplasmic reticulum (ER) adapter protein (encoded by TMEM173 gene) that causes chronic inflammation [93][87]. The nucleic acid pattern recognition receptor (cyclic GMP-AMP synthase, cGAS) is the upstream protein that activates STING. It has been found that cGAS can be localized on the cell membrane, in the cytoplasm, and in the nucleus. cGAS binds to double-stranded DNA of exogenous and endogenous origin (including mtDNA) [94][88]. The binding of cGAS to mtDNA induces the synthesis of cyclic dinucleotide (CDN)-2′,3′-cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). The synthesized cGAMP binds to the active pocket site STING and activates further signal transduction [95][89].
cGAS-dependent and -independent (ER stress) stimulation of STING leads to subsequent transport and activation of TANK-binding kinase 1 (TNK1) for phosphorylation of the regulatory factor IFN 3 (IRF3). IRF3 then dissociates and dimerizes and becomes transcriptionally active. IRF3 then migrates to the nucleus and triggers an IRF3-dependent IFN-1 response [96][90]. In addition, TNK1 induces nuclear transfer of nuclear factor kappa B (NF-kB), which triggers the transcription of pro-inflammatory cytokines [97][91].
In addition, the cGAS-STING-TBK1 pathway, which involves IKK kinases [98][92], promotes the translocation of nuclear factor-kappa B (NF-kB) to the nucleus [99][93], which also regulates the transcription of pro-inflammatory factors. STING has been shown to induce canonical/noncanonical activation of the NF-kB pathway. Thus, activation of STING (via cGAS) stimulates NF-kB and mitogen-activated protein kinase signaling via TNK1 [97][91].
In vitro/in vivo studies have shown that TNK1 is essential for STING-induced NF-kB activation and downstream STING kinases (TBK1, IkB kinase ε-IKKε) act redundantly to mediate stimulation of STING-NF-Kb, which triggers transcription of pro-inflammatory cytokines [97,100][91][94].
In addition to the inflammatory response, STING also regulates the process of autophagy/mitophagy, lytic cell death, apoptosis, etc. In this regard, these signaling outputs may or may not depend on upstream cGAS and second messenger cGAMP [101][95].
It was found that in the background of MetS there is a violation of mitophagy processes responsible for the removal of damaged mitochondria. In addition, metabolic diseases are associated with a decrease in mtDNA regulators and changes in mitochondrial integrity and structure. This contributes to mitochondrial stress and mtDNA release, leading to inflammatory responses associated with obesity and MetS [102][96].
Studies have shown that high levels of mtDNA fragments in plasma and mtDNA damage are positively correlated with chronic inflammation and oxidative stress [103,104][97][98]. This suggests an important role of free mtDNA as DAMPs and activators of the cGAS-STING-TBK1 signaling cascade.
TFAM heterogeneity was found to promote the release of mtDNA into the cytosol and maintain cGAS. Thus, mtDNA in the cytosol can activate the cGAS-STING pathway and increase the expression of interferon-stimulated genes (ISG) through IFN-1 signaling [105][99].
Studies of adipose tissue in an animal model (mouse) fed a high-fat diet (HFD) and in obese humans have shown high expression of the transcription factor IRF3, which is associated with inflammation, macrophage infiltration, and insulin resistance [106][100]. In addition, Bai’s research group [107][101] investigated the mechanism of activation of the cGAS-cGAMP-STING signaling pathway in obesity that leads to insulin resistance, sterile inflammation, and energy dysregulation. Adipose tissue shows overexpression of an oxidoreductase-like protein with a disulfide bond (DsbA-L). DsbA-L is a mitochondrial matrix protein that is also localized to the endoplasmic reticulum (ER) and has a chaperone function in adiponectin multimerization and is involved in the folding of key mitochondrial proteins critical for mtDNA replication [108][102]. Obesity suppresses the expression of DsbA-L and destabilizes mtDNA, promoting its release and activation of the cGAS-STING pathway, which increases the expression of TBK1, NF-kB, P65, and IRF3 genes. Increased levels of TBK1, NF-kB, P65, and IRF3 suppress insulin signaling and promote the inflammatory response.
Moreover, phosphorylated TBK1 negatively regulates thermogenesis and energy expenditure by activating phosphodiesterase-PDE3B/PDE4 to suppress protein kinase A (PKA) signaling in adipocytes and HFD-treated mice [107,109][101][103]. Simultaneously, overexpression of DsbA-L protected against mtDNA-induced stimulation of the cGAS-STING pathway and suppressed inflammatory factors via a mechanism unrelated to adiponectin and ER localization. Overexpression of DsbA-L restored PKA signaling and increased energy expenditure [107,109][101][103]. Moreover, HFD-induced obesity activates TBK1 kinase and deregulates energy homeostasis by directly inhibiting AMPK at Thr142 of the AMPKα subunit in adipose tissue [110][104]. At the same time, TBK1 has bidirectional activity against the inflammatory response in adipocytes [107,110][101][104]. In this study, obesity was found to trigger the release of mtDNA and cause activation of a signaling pathway.
Therefore, a more detailed investigation of the molecular aspect of mtDNA-mediated activation and the cGAS-STING-IRF3 pathway in the development and progression of the MetS will allow the identification of specific targets for effective regulation of recovery processes and reduce the risk of developing severe pathologies.
7.2. TLR9
Toll-like receptor 9 (TLR9) is an intracellular pattern recognition receptor and is expressed in immune cells (dendritic cells, B lymphocytes, NK cells, macrophages, etc.) and non-immune cells (cardiomyocytes, muscle, and endothelial cells) [98,111][92][105].
Mitochondrially released mtDNA interacts with TLR9 against a background of oxidative stress and decreased autophagy and promotes an inflammatory response (in mice) [112][106]. It was found that mtDNA is taken up by immune cells in which it triggers an inflammatory response by activating TLR9 [113][107]. Activation of TLR9 via unmethylated mtDNA-CpG motifs leads to the induction of NF-kB and MAPK signaling cascades [89,111][83][105].
Interestingly, mtDNA-bound TFAM can interact with the plasma membrane receptor RAGE and induce internalization of mtDNA, facilitating its recognition by TLR9 [98][92].
TLR9 is known to signal through myeloid differentiation protein 88 (MyD88), which activates kinases and transcription factors: MAPK, NF-kB, IRF7, to enhance proinflammatory processes and type I interferon response. Studies have shown that full-length TLR9 is translocated through the Golgi apparatus and endolysosomal compartment under the guidance of the helper protein UNC93B1 and then cleaved to acquire the ability to take up DNA [114][108]. The recruitment of TLR9 to the endolysosome is a key moment for the formation of its proteolytic activity [98][92].
In peripheral blood monocytes from patients with MetS, increased expression of endosomal TLR9 was found compared with controls, which correlated with increased nuclear expression of NF-kB [115][109].
In addition, work in Tlr9-/- mice showed a lower inflammatory response compared to wild-type mice fed a high-calorie diet and demonstrated the relationship between adipocyte degeneration, free DNA release, TLR9 activation, and the development of adipose tissue inflammation in an obesity background [116][110].
In a recent study, patients with MetS were found to have increased levels of oxidized mtDNA (ox-mtDNA) in plasma and increased TLR9 expression in PBMCs. At the same time, rats receiving HFD also had increased levels of ox-mtDNA expression in plasma and TLR9 expression in cardiac immune cells. Interestingly, in patients with MetS, an increase in circulating ox-mtDNA activated the TLR9-NF-kB pathway and stimulated secretion of IL-1β, IL-6, and IL-8 (a block of culture work on THP-1 cells) [117][111].
Thus, the broad spectrum of activity of the activated TLR9 pathway under the influence of mtDNA suggests its key role in the pathogenesis of MetS and may serve as a promising target whose regulation will reduce the negative consequences of the disease.
7.3. Pyrin Domain of NLR Family-Containing Inflammasome 3 (NLRP3)
Inflammasomes fall into several categories: (1) nucleotide-binding domain (NOD); leucine-rich repeats (LRR), containing protein (NLR), or NLR inflammasomes; (2) absent in melanoma 2 (AIM2) and pyrin inflammasomes; (3) non-canonical inflammasomes [118][112]. mtDNA is recognized by two members of the immunogenic receptor superfamily: AIM2 and NRLP3. NRLP3 inflammasomes are cytoplasmic multiprotein complexes of the innate immune system [81][75]. NRLP3 inflammasomes are localized in neutrophils, monocytes, dendritic cells, macrophages, and nonhematopoietic cells [119][113].
The inflammasome is activated by PAMPs and DAMPs (including mtDNA). The inflammasome is activated by two sequential signals. First, NF-kB is stimulated (via TLR9, TLR4, TLR7), which triggers the inflammatory response and prepares the cell for further activation. The second signal is a trigger that interacts with the recognition part of the complex and leads to oligomerization/activation of the inflammasome particle. The specific inflammasome protein is the recognition protein and the common one is the scaffold adaptor protein ASC that relays the caspase-1 signal. Assembly and oligomerization of the inflammasome lead to cleavage of caspase-1 and trigger secretion of IL-1β, whose expression was triggered by the start signal [98][92].
When NRLP3 is activated, the caspase 1 subunit of the NRLP3 complex cleaves pro-interleukins into mature IL-1β, IL-18, key markers of mild inflammation. NRLP3 is considered a key factor responsible for the initiation and progression of chronic inflammation [5][19].
Obesity, diabetes, and MetS have been shown to be associated with increased NRLP3 activity in subcutaneous adipose tissue (SAT). At the same time, inflammatory mediators, IL-1β, IL-18, and caspase-1 levels at SAT are significantly increased in patients with MetS compared to controls, which may contribute to an increase in insulin resistance, inflammation, and SAT fibrosis [120][114].
In addition, disruption of NRLP3 in adipose tissue decreases levels of pro-inflammatory cytokines and restores insulin sensitivity in obese mice [121][115].
An interaction between mtDNA and NLRP3 has been described for autophagic protein-deficient mouse macrophages (LC3B, beclin1). Against the background of LC3B/beclin1 deficiency, accumulation of dysfunctional mitochondria and NLRP3/ROS-dependent mtDNA translocation to the cytosol were observed. Cytosolic mtDNA was involved in the development of the inflammatory response through the activation of IL-1β/IL-18 secretion [122][116]. Studies have also shown that autophagic clearance of damaged mitochondria (and mtDNA) prevents the development of inflammation. In addition, the compound 8-OH-dG in ox-mtDNA has been suggested to play an important role in NLRP3 activation [123][117].
Thus, MetS is directly related to the development of chronic, noninfectious (sterile) inflammation associated with mitochondrial damage and the release of mtDNA into the cytoplasm, which activates various signaling pathways (Figure 1).
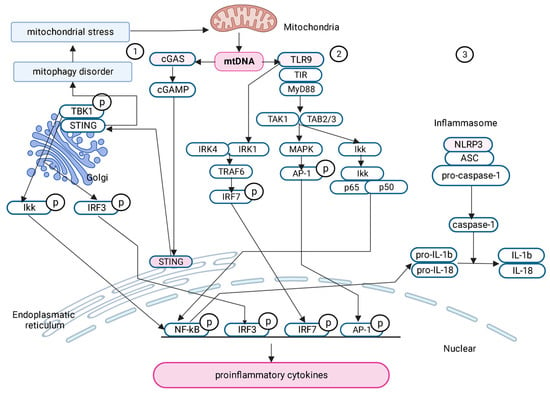
Figure 1. Signaling pathways activated by free mtDNA in the pathogenesis of metabolic syndrome. (1) cGAS- STING signaling pathway; (2) TLR9 signaling pathway; (3) NLRP3 signaling pathway.
Gradually, some aspects of the molecular action of mtDNA in the development of metabolic diseases are becoming better understood, but many questions remain unexplored and need to be investigated in depth.
References
- Hosgood, H.D.; Liu, C.-S.; Rothman, N.; Weinstein, S.J.; Bonner, M.R.; Shen, M.; Lim, U.; Virtamo, J.; Cheng, W.; Albanes, D.; et al. Mitochondrial DNA Copy Number and Lung Cancer Risk in a Prospective Cohort Study. Carcinogenesis 2010, 31, 847–849.
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep. 2020, 21, e49799.
- Dashti, M.; Alsaleh, H.; Eaaswarkhanth, M.; John, S.E.; Nizam, R.; Melhem, M.; Hebbar, P.; Sharma, P.; Al-Mulla, F.; Thanaraj, T.A. Delineation of Mitochondrial DNA Variants From Exome Sequencing Data and Association of Haplogroups With Obesity in Kuwait. Front. Genet. 2021, 12, 626260.
- Ding, X.; Fang, T.; Pang, X.; Pan, X.; Tong, A.; Lin, Z.; Zheng, S.; Zheng, N. Mitochondrial DNA Abnormalities and Metabolic Syndrome. Front. Cell Dev. Biol. 2023, 11, 1153174.
- Révész, D.; Verhoeven, J.E.; Picard, M.; Lin, J.; Sidney, S.; Epel, E.S.; Penninx, B.W.J.H.; Puterman, E. Associations Between Cellular Aging Markers and Metabolic Syndrome: Findings From the CARDIA Study. J. Clin. Endocrinol. Metab. 2018, 103, 148–157.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462.
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking Outside the Nucleus: Mitochondrial DNA Copy Number in Health and Disease. Mitochondrion 2020, 53, 214–223.
- Guha, M.; Avadhani, N.G. Mitochondrial Retrograde Signaling at the Crossroads of Tumor Bioenergetics, Genetics and Epigenetics. Mitochondrion 2013, 13, 577–591.
- Jeng, J.-Y.; Yeh, T.-S.; Lee, J.-W.; Lin, S.-H.; Fong, T.-H.; Hsieh, R.-H. Maintenance of Mitochondrial DNA Copy Number and Expression Are Essential for Preservation of Mitochondrial Function and Cell Growth. J. Cell. Biochem. 2008, 103, 347–357.
- Malik, A.N.; Czajka, A. Is Mitochondrial DNA Content a Potential Biomarker of Mitochondrial Dysfunction? Mitochondrion 2013, 13, 481–492.
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial Biogenesis in the Metabolic Syndrome and Cardiovascular Disease. J. Mol. Med. 2010, 88, 993–1001.
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414.
- Liu, J.; Lloyd, S.G. High-Fat, Low-Carbohydrate Diet Alters Myocardial Oxidative Stress and Impairs Recovery of Cardiac Function after Ischemia and Reperfusion in Obese Rats. Nutr. Res. 2013, 33, 311–321.
- Skuratovskaia, D.; Zatolokin, P.; Vulf, M.; Mazunin, I.; Litvinova, L. Interrelation of Chemerin and TNF-α with MtDNA Copy Number in Adipose Tissues and Blood Cells in Obese Patients with and without Type 2 Diabetes. BMC Med. Genom. 2019, 12, 40.
- Di Rienzo, M.; Romagnoli, A.; Ciccosanti, F.; Refolo, G.; Consalvi, V.; Arena, G.; Valente, E.M.; Piacentini, M.; Fimia, G.M. AMBRA1 Regulates Mitophagy by Interacting with ATAD3A and Promoting PINK1 Stability. Autophagy 2022, 18, 1752–1762.
- Meng, S.; Wu, S.; Liang, L.; Liang, G.; Giovannucci, E.; De Vivo, I.; Nan, H. Leukocyte Mitochondrial DNA Copy Number, Anthropometric Indices, and Weight Change in US Women. Oncotarget 2016, 7, 60676–60686.
- Skuratovskaia, D.A.; Sofronova, J.K.; Zatolokin, P.A.; Popadin, K.Y.; Vasilenko, M.A.; Litvinova, L.S.; Mazunin, I.O. Additional Evidence of the Link between MtDNA Copy Number and the Body Mass Index. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2018, 29, 1240–1244.
- Liu, X.; Longchamps, R.J.; Wiggins, K.L.; Raffield, L.M.; Bielak, L.F.; Zhao, W.; Pitsillides, A.; Blackwell, T.W.; Yao, J.; Guo, X.; et al. Association of Mitochondrial DNA Copy Number with Cardiometabolic Diseases. Cell Genom. 2021, 1, 100006.
- Ravaut, G.; Légiot, A.; Bergeron, K.-F.; Mounier, C. Monounsaturated Fatty Acids in Obesity-Related Inflammation. Int. J. Mol. Sci. 2020, 22, 330.
- Koller, A.; Fazzini, F.; Lamina, C.; Rantner, B.; Kollerits, B.; Stadler, M.; Klein-Weigel, P.; Fraedrich, G.; Kronenberg, F. Mitochondrial DNA Copy Number Is Associated with All-Cause Mortality and Cardiovascular Events in Patients with Peripheral Arterial Disease. J. Intern. Med. 2020, 287, 569–579.
- Agius, R.; Pace, N.P.; Fava, S. Reduced Leukocyte Mitochondrial Copy Number in Metabolic Syndrome and Metabolically Healthy Obesity. Front. Endocrinol. 2022, 13, 886957.
- Huang, C.-H.; Su, S.-L.; Hsieh, M.-C.; Cheng, W.-L.; Chang, C.-C.; Wu, H.-L.; Kuo, C.-L.; Lin, T.-T.; Liu, C.-S. Depleted Leukocyte Mitochondrial DNA Copy Number in Metabolic Syndrome. J. Atheroscler. Thromb. 2011, 18, 867–873.
- Kim, J.-H.; Im, J.-A.; Lee, D.-C. The Relationship between Leukocyte Mitochondrial DNA Contents and Metabolic Syndrome in Postmenopausal Women. Menopause 2012, 19, 582–587.
- Fazzini, F.; Lamina, C.; Raftopoulou, A.; Koller, A.; Fuchsberger, C.; Pattaro, C.; Del Greco, F.M.; Döttelmayer, P.; Fendt, L.; Fritz, J.; et al. Association of Mitochondrial DNA Copy Number with Metabolic Syndrome and Type 2 Diabetes in 14 176 Individuals. J. Intern. Med. 2021, 290, 190–202.
- Mengel-From, J.; Thinggaard, M.; Dalgård, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA Copy Number in Peripheral Blood Cells Declines with Age and Is Associated with General Health among Elderly. Hum. Genet. 2014, 133, 1149–1159.
- Kim, J.-H.; Kim, H.K.; Ko, J.-H.; Bang, H.; Lee, D.-C. The Relationship between Leukocyte Mitochondrial DNA Copy Number and Telomere Length in Community-Dwelling Elderly Women. PLoS ONE 2013, 8, e67227.
- Cree, L.M.; Patel, S.K.; Pyle, A.; Lynn, S.; Turnbull, D.M.; Chinnery, P.F.; Walker, M. Age-Related Decline in Mitochondrial DNA Copy Number in Isolated Human Pancreatic Islets. Diabetologia 2008, 51, 1440–1443.
- Picard, M.; Turnbull, D.M. Linking the Metabolic State and Mitochondrial DNA in Chronic Disease, Health, and Aging. Diabetes 2013, 62, 672–678.
- Memon, A.A.; Sundquist, J.; Hedelius, A.; Palmér, K.; Wang, X.; Sundquist, K. Association of Mitochondrial DNA Copy Number with Prevalent and Incident Type 2 Diabetes in Women: A Population-Based Follow-up Study. Sci. Rep. 2021, 11, 4608.
- Wang, J.; Liang, H.; Huang, R.; Weng, X.; Zheng, L.; Wang, Y.; Zheng, X.; Gu, Z.; Chen, F.; Shao, J.; et al. Higher Mitochondrial DNA Copy Number Is Associated with Metformin-Induced Weight Loss. Commun. Med. 2023, 3, 29.
- Skuratovskaia, D.; Litvinova, L.; Vulf, M.; Zatolokin, P.; Popadin, K.; Mazunin, I. From Normal to Obesity and Back: The Associations between Mitochondrial DNA Copy Number, Gender, and Body Mass Index. Cells 2019, 8, 430.
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463.
- Castegna, A.; Iacobazzi, V.; Infantino, V. The Mitochondrial Side of Epigenetics. Physiol. Genom. 2015, 47, 299–307.
- D’Aquila, P.; Bellizzi, D.; Passarino, G. Mitochondria in Health, Aging and Diseases: The Epigenetic Perspective. Biogerontology 2015, 16, 569–585.
- Castellani, C.A.; Longchamps, R.J.; Sumpter, J.A.; Newcomb, C.E.; Lane, J.A.; Grove, M.L.; Bressler, J.; Brody, J.A.; Floyd, J.S.; Bartz, T.M.; et al. Mitochondrial DNA Copy Number Can Influence Mortality and Cardiovascular Disease via Methylation of Nuclear DNA CpGs. Genome Med. 2020, 12, 84.
- Murphy, S.K.; Huang, Z.; Hoyo, C. Differentially Methylated Regions of Imprinted Genes in Prenatal, Perinatal and Postnatal Human Tissues. PLoS ONE 2012, 7, e40924.
- Fradin, D.; Boëlle, P.-Y.; Belot, M.-P.; Lachaux, F.; Tost, J.; Besse, C.; Deleuze, J.-F.; De Filippo, G.; Bougnères, P. Genome-Wide Methylation Analysis Identifies Specific Epigenetic Marks In Severely Obese Children. Sci. Rep. 2017, 7, 46311.
- Dou, X.; Boyd-Kirkup, J.D.; McDermott, J.; Zhang, X.; Li, F.; Rong, B.; Zhang, R.; Miao, B.; Chen, P.; Cheng, H.; et al. The Stand-Biased Mitochondrial DNA Methylome And Its Regulation By DNMT3A. Genome Res. 2019, 10, 1622–1634.
- Guantes, R.; Rastrojo, A.; Neves, R.; Lima, A.; Aguado, B.; Iborra, F.J. Global Variability in Gene Expression and Alternative Splicing Is Modulated by Mitochondrial Content. Genome Res. 2015, 25, 633–644.
- Bai, Y.; Guo, Z.; Xu, J.; Zhang, J.; Cui, L.; Zhang, H.; Zang, S.; Ai, X. Single Nucleotide Polymorphisms In The D-Loop Region Of Mitochondrial DNA And Age-At0Onset Of Patients With Chronic Kidney Disease. Chin. Med. J. 2014, 127, 3088–3091.
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced Mitochondrial D-Loop Methylation Levels in Sporadic Amyotrophic Lateral Sclerosis. Clin. Epigenetics 2020, 12, 137.
- Mposhi, A.; Van der Wijst, M.G.; Faber, K.N.; Rots, M.G. Regulation of mitochondrial gene expression, the epigenetic enigma. Front. Biosci. (Landmark Ed) 2017, 22, 1099–1113.
- Dostal, V.; Churchill, M.E.A. Cytosine Methylation of Mitochondrial DNA at CpG Sequences Impacts Transcription Factor A DNA Binding and Transcription. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 598–607.
- Gao, J.; Wen, S.; Zhou, H.; Feng, S. De-Methylation of Displacement Loop of Mitochondrial DNA Is Associated with Increased Mitochondrial Copy Number and Nicotinamide Adenine Dinucleotide Subunit 2 Expression in Colorectal Cancer. Mol. Med. Rep. 2015, 12, 7033–7038.
- Park, S.H.; Lee, S.Y.; Kim, S.A. Mitochondrial DNA Methylation Is Higher in Acute Coronary Syndrome Than in Stable Coronary Artery Disease. In Vivo 2021, 35, 181–189.
- Low, H.C.; Chilian, W.M.; Ratnam, W.; Karupaiah, T.; Md Noh, M.F.; Mansor, F.; Ng, Z.X.; Pung, Y.F. Changes in Mitochondrial Epigenome in Type 2 Diabetes Mellitus. Br. J. Biomed. Sci. 2023, 80, 10884.
- Hao, Z.; Wu, T.; Cui, X.; Zhu, P.; Tan, C.; Dou, X.; Hsu, K.W.; Lin, Y.T.; Peng, P.H.; Zang, L.S.; et al. N6-Deoxyadenosine Methylation In Mammalian Mitochondrial DNA. Mol. Cell 2020, 78, 382–395.
- Bordoni, L.; Smerilli, V.; Nasuti, C.; Gabbianelli, R. Mitochondrial DNA Methylation and Copy Number Predict Body Composition in a Young Female Population. J. Transl. Med. 2019, 17, 399.
- Bordoni, L.; Perugini, J.; Petracci, I.; Mercurio, E.D.; Lezoche, G.; Guerrieri, M.; Giordano, A.; Gabbianelli, R. Mitochondrial DNA in Visceral Adipose Tissue in Severe Obesity: From Copy Number to D-Loop Methylation. Front. Biosci. (Landmark Ed) 2022, 27, 172.
- Corsi, S.; Iodice, S.; Vigna, L.; Cayir, A.; Mathers, J.C.; Bollati, V.; Byun, H.-M. Platelet Mitochondrial DNA Methylation Predicts Future Cardiovascular Outcome in Adults with Overweight and Obesity. Clin. Epigenetics 2020, 12, 29.
- Baccarelli, A.A.; Byun, H.-M. Platelet Mitochondrial DNA Methylation: A Potential New Marker of Cardiovascular Disease. Clin. Epigenetics 2015, 7, 44.
- Zheng, L.D.; Linarelli, L.E.; Liu, L.; Wall, S.S.; Greenawald, M.H.; Seidel, R.W.; Estabrooks, P.A.; Almeida, F.A.; Cheng, Z. Insulin Resistance Is Associated with Epigenetic and Genetic Regulation of Mitochondrial DNA in Obese Humans. Clin. Epigenetics 2015, 7, 60.
- Khotina, V.A.; Vinokurov, A.Y.; Bagheri Ekta, M.; Sukhorukov, V.N.; Orekhov, A.N. Creation Of Mitochondrial Disease Models Using Mitochondrial DNA Editing. Biomrdicines 2023, 2, 532.
- Katada, S.; Mito, T.; Ogasawara, E.; Hayashi, J.-I.; Nakada, K. Mitochondrial DNA with a Large-Scale Deletion Causes Two Distinct Mitochondrial Disease Phenotypes in Mice. G3 (Bethesda) 2013, 3, 1545–1552.
- Farmer, T.; Naslavsky, N.; Caplan, S. Tying Trafficking to Fusion and Fission at the Mighty Mitochondria. Traffic 2018, 19, 569–577.
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407.
- Palmieri, V.O.; De Rasmo, D.; Signorile, A.; Sardanelli, A.M.; Grattagliano, I.; Minerva, F.; Cardinale, G.; Portincasa, P.; Papa, S.; Palasciano, G. T16189C Mitochondrial DNA Variant Is Associated with Metabolic Syndrome in Caucasian Subjects. Nutrition 2011, 27, 773–777.
- Mueller, E.E.; Eder, W.; Ebner, S.; Schwaiger, E.; Santic, D.; Kreindl, T.; Stanger, O.; Paulweber, B.; Iglseder, B.; Oberkofler, H.; et al. The Mitochondrial T16189C Polymorphism Is Associated with Coronary Artery Disease in Middle European Populations. PLoS ONE 2011, 6, e16455.
- Maassen, J.A.; ’T Hart, L.M.; Van Essen, E.; Heine, R.J.; Nijpels, G.; Jahangir Tafrechi, R.S.; Raap, A.K.; Janssen, G.M.C.; Lemkes, H.H.P.J. Mitochondrial Diabetes: Molecular Mechanisms and Clinical Presentation. Diabetes 2004, 53 (Suppl. S1), S103–S109.
- Park, H.; Davidson, E.; King, M.P. The Pathogenic A3243G Mutation in Human Mitochondrial TRNALeu(UUR) Decreases the Efficiency of Aminoacylation. Biochemistry 2003, 42, 958–964.
- Ye, W.; Chen, S.; Jin, S.; Lu, J. A Novel Heteroplasmic Mitochondrial DNA Mutation, A8890G, in a Patient with Juvenile-onset Metabolic Syndrome: A Case Report. Mol. Med. Rep. 2013, 8, 1060–1066.
- Wallace, D.C. Mitochondrial DNA Variation in Human Radiation and Disease. Cell 2015, 163, 33–38.
- Ebner, S.; Mangge, H.; Langhof, H.; Halle, M.; Siegrist, M.; Aigner, E.; Paulmichl, K.; Paulweber, B.; Datz, C.; Sperl, W.; et al. Mitochondrial Haplogroup T Is Associated with Obesity in Austrian Juveniles and Adults. PLoS ONE 2015, 10, e0135622.
- Nardelli, C.; Labruna, G.; Liguori, R.; Mazzaccara, C.; Ferrigno, M.; Capobianco, V.; Pezzuti, M.; Castaldo, G.; Farinaro, E.; Contaldo, F.; et al. Haplogroup T Is an Obesity Risk Factor: Mitochondrial DNA Haplotyping in a Morbid Obese Population from Southern Italy. Biomed. Res. Int. 2013, 2013, 631082.
- Dashti, M.; Alsaleh, H.; Rodriguez-Flores, J.L.; Eaaswarkhanth, M.; Al-Mulla, F.; Thanaraj, T.A. Mitochondrial Haplogroup J Associated with Higher Risk of Obesity in the Qatari Population. Sci. Rep. 2021, 11, 1091.
- Chalkia, D.; Chang, Y.-C.; Derbeneva, O.; Lvova, M.; Wang, P.; Mishmar, D.; Liu, X.; Singh, L.N.; Chuang, L.-M.; Wallace, D.C. Mitochondrial DNA Associations with East Asian Metabolic Syndrome. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 878–892.
- Fuku, N.; Park, K.S.; Yamada, Y.; Nishigaki, Y.; Cho, Y.M.; Matsuo, H.; Segawa, T.; Watanabe, S.; Kato, K.; Yokoi, K.; et al. Mitochondrial Haplogroup N9a Confers Resistance against Type 2 Diabetes in Asians. Am. J. Hum. Genet. 2007, 80, 407–415.
- Guo, L.-J.; Oshida, Y.; Fuku, N.; Takeyasu, T.; Fujita, Y.; Kurata, M.; Sato, Y.; Ito, M.; Tanaka, M. Mitochondrial Genome Polymorphisms Associated with Type-2 Diabetes or Obesity. Mitochondrion 2005, 5, 15–33.
- Fang, H.; Hu, N.; Zhao, Q.; Wang, B.; Zhou, H.; Fu, Q.; Shen, L.; Chen, X.; Shen, F.; Lyu, J. MtDNA Haplogroup N9a Increases the Risk of Type 2 Diabetes by Altering Mitochondrial Function and Intracellular Mitochondrial Signals. Diabetes 2018, 67, 1441–1453.
- Liao, W.-Q.; Pang, Y.; Yu, C.-A.; Wen, J.-Y.; Zhang, Y.-G.; Li, X.-H. Novel Mutations of Mitochondrial DNA Associated with Type 2 Diabetes in Chinese Han Population. Tohoku J. Exp. Med. 2008, 215, 377–384.
- Niu, Q.; Zhang, W.; Wang, H.; Guan, X.; Lu, J.; Li, W. Effects of Mitochondrial Haplogroup N9a on Type 2 Diabetes Mellitus and Its Associated Complications. Exp. Ther. Med. 2015, 10, 1918–1924.
- Loo, J.-H.; Trejaut, J.A.; Yen, J.-C.; Chen, Z.-S.; Ng, W.-M.; Huang, C.-Y.; Hsu, K.-N.; Hung, K.-H.; Hsiao, Y.; Wei, Y.-H.; et al. Mitochondrial DNA Association Study of Type 2 Diabetes with or without Ischemic Stroke in Taiwan. BMC Res. Notes 2014, 7, 223.
- Shen, F.-C.; Weng, S.-W.; Tsai, M.-H.; Su, Y.-J.; Li, S.-C.; Chang, S.-J.; Chen, J.-F.; Chang, Y.-H.; Liou, C.-W.; Lin, T.-K.; et al. Mitochondrial Haplogroups Have a Better Correlation to Insulin Requirement than Nuclear Genetic Variants for Type 2 Diabetes Mellitus in Taiwanese Individuals. J. Diabetes Investig. 2022, 13, 201–208.
- Jiang, W.; Li, R.; Zhang, Y.; Wang, P.; Wu, T.; Lin, J.; Yu, J.; Gu, M. Mitochondrial DNA Mutations Associated with Type 2 Diabetes Mellitus in Chinese Uyghur Population. Sci. Rep. 2017, 7, 16989.
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of MtDNA-Mediated Inflammation. Cells 2021, 10, 2898.
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously Oxidized Mitochondrial DNA Induces in Vivo and in Vitro Inflammatory Responses. J. Leukoc. Biol. 2004, 75, 995–1000.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414.
- Pazmandi, K.; Agod, Z.; Kumar, B.V.; Szabo, A.; Fekete, T.; Sogor, V.; Veres, A.; Boldogh, I.; Rajnavolgyi, E.; Lanyi, A.; et al. Oxidative Modification Enhances the Immunostimulatory Effects of Extracellular Mitochondrial DNA on Plasmacytoid Dendritic Cells. Free. Radic. Biol. Med. 2014, 77, 281–290.
- Kunkel, G.H.; Chaturvedi, P.; Tyagi, S.C. Mitochondrial Pathways to Cardiac Recovery: TFAM. Heart Fail Rev. 2016, 21, 499–517.
- Piantadosi, C.A. Mitochondrial DNA, Oxidants, and Innate Immunity. Free. Radic. Biol. Med. 2020, 152, 455–461.
- Wang, L.; Zhang, Q.; Yuan, K.; Yuan, J. MtDNA in the Pathogenesis of Cardiovascular Diseases. Dis. Markers 2021, 2021, 7157109.
- Krychtiuk, K.A.; Wurm, R.; Ruhittel, S.; Lenz, M.; Huber, K.; Wojta, J.; Heinz, G.; Hülsmann, M.; Speidl, W.S. Release of Mitochondrial DNA Is Associated with Mortality in Severe Acute Heart Failure. Eur. Heart J. Acute Cardiovasc. Care 2020, 9, 419–428.
- Bliksøen, M.; Mariero, L.H.; Torp, M.K.; Baysa, A.; Ytrehus, K.; Haugen, F.; Seljeflot, I.; Vaage, J.; Valen, G.; Stensløkken, K.-O. Extracellular MtDNA Activates NF-ΚB via Toll-like Receptor 9 and Induces Cell Death in Cardiomyocytes. Basic Res. Cardiol. 2016, 111, 42.
- Donath, M.Y.; Meier, D.T.; Böni-Schnetzler, M. Inflammation in the Pathophysiology and Therapy of Cardiometabolic Disease. Endocr. Rev. 2019, 40, 1080–1091.
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating Mitochondrial DNA Increases with Age and Is a Familiar Trait: Implications for “Inflamm-Aging. ” Eur. J. Immunol. 2014, 44, 1552–1562.
- Ueda, H.; Yamaguchi, O.; Taneike, M.; Akazawa, Y.; Wada-Kobayashi, H.; Sugihara, R.; Yorifuji, H.; Nakayama, H.; Omiya, S.; Murakawa, T.; et al. Administration of a TLR9 Inhibitor Attenuates the Development and Progression of Heart Failure in Mice. JACC Basic Transl. Sci. 2019, 4, 348–363.
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med. 2014, 371, 507–518.
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557.
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791.
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398.
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328–5341.
- Rodríguez-Nuevo, A.; Zorzano, A. The Sensing of Mitochondrial DAMPs by Non-Immune Cells. Cell Stress 2019, 3, 195–207.
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630.
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-ΚB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492.
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of CGAS-STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521.
- Oduro, P.K.; Zheng, X.; Wei, J.; Yang, Y.; Wang, Y.; Zhang, H.; Liu, E.; Gao, X.; Du, M.; Wang, Q. The CGAS-STING Signaling in Cardiovascular and Metabolic Diseases: Future Novel Target Option for Pharmacotherapy. Acta Pharm. Sin. B 2022, 12, 50–75.
- Yuzefovych, L.V.; Pastukh, V.M.; Ruchko, M.V.; Simmons, J.D.; Richards, W.O.; Rachek, L.I. Plasma Mitochondrial DNA Is Elevated in Obese Type 2 Diabetes Mellitus Patients and Correlates Positively with Insulin Resistance. PLoS ONE 2019, 14, e0222278.
- Nie, S.; Lu, J.; Wang, L.; Gao, M. Pro-Inflammatory Role of Cell-Free Mitochondrial DNA in Cardiovascular Diseases. IUBMB Life 2020, 72, 1879–1890.
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. CGAS Senses Long and HMGB/TFAM-Bound U-Turn DNA by Forming Protein-DNA Ladders. Nature 2017, 549, 394–398.
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 Promotes Adipose Inflammation and Insulin Resistance and Represses Browning. J. Clin. Investig. 2016, 126, 2839–2854.
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L Prevents Obesity-Induced Inflammation and Insulin Resistance by Suppressing the MtDNA Release-Activated CGAS-CGAMP-STING Pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201.
- Zhou, L.; Liu, M.; Zhang, J.; Chen, H.; Dong, L.Q.; Liu, F. DsbA-L Alleviates Endoplasmic Reticulum Stress-Induced Adiponectin Downregulation. Diabetes 2010, 59, 2809–2816.
- Bai, J.; Cervantes, C.; He, S.; He, J.; Plasko, G.R.; Wen, J.; Li, Z.; Yin, D.; Zhang, C.; Liu, M.; et al. Mitochondrial Stress-Activated CGAS-STING Pathway Inhibits Thermogenic Program and Contributes to Overnutrition-Induced Obesity in Mice. Commun. Biol. 2020, 3, 257.
- Zhao, P.; Wong, K.I.; Sun, X.; Reilly, S.M.; Uhm, M.; Liao, Z.; Skorobogatko, Y.; Saltiel, A.R. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell 2018, 172, 731–743.e12.
- Guo, Z.; Tang, N.; Liu, F.-Y.; Yang, Z.; Ma, S.-Q.; An, P.; Wu, H.-M.; Fan, D.; Tang, Q.-Z. TLR9 Deficiency Alleviates Doxorubicin-Induced Cardiotoxicity via the Regulation of Autophagy. J. Cell. Mol. Med. 2020, 24, 10913–10923.
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA That Escapes from Autophagy Causes Inflammation and Heart Failure. Nature 2012, 485, 251–255.
- Xie, L.; He, S.; Kong, N.; Zhu, Y.; Tang, Y.; Li, J.; Liu, Z.; Liu, J.; Gong, J. Cpg-ODN, a TLR9 Agonist, Aggravates Myocardial Ischemia/Reperfusion Injury by Activation of TLR9-P38 MAPK Signaling. Cell. Physiol. Biochem. 2018, 47, 1389–1398.
- Pelka, K.; Phulphagar, K.; Zimmermann, J.; Stahl, R.; Schmid-Burgk, J.L.; Schmidt, T.; Spille, J.-H.; Labzin, L.I.; Agrawal, S.; Kandimalla, E.R.; et al. Cutting Edge: The UNC93B1 Tyrosine-Based Motif Regulates Trafficking and TLR Responses via Separate Mechanisms. J. Immunol. 2014, 193, 3257–3261.
- Devaraj, S.; Adams-Huet, B.; Jialal, I. Endosomal Toll-Like Receptor Status in Patients with Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2015, 13, 477–480.
- Nishimoto, S.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Murata, C.; Kim-Kaneyama, J.R.; Sato, F.; Bando, M.; Yagi, S.; et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci. Adv. 2016, 2, e1501332.
- Ye, W.; Wen, C.; Zeng, A.; Hu, X. Increased Levels of Circulating Oxidized Mitochondrial DNA Contribute to Chronic Inflammation in Metabolic Syndrome, and MitoQ-Based Antioxidant Therapy Alleviates This DNA-Induced Inflammation. Mol. Cell. Endocrinol. 2023, 560, 111812.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Qiu, Z.; He, Y.; Ming, H.; Lei, S.; Leng, Y.; Xia, Z.-Y. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J. Diabetes Res. 2019, 2019, 8151836.
- Pahwa, R.; Singh, A.; Adams-Huet, B.; Devaraj, S.; Jialal, I. Increased Inflammasome Activity in Subcutaneous Adipose Tissue of Patients with Metabolic Syndrome. Diabetes/Metabolism Res. Rev. 2021, 37, e3383.
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188.
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230.
- Ward, G.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; Meyer, B.S.; McLemore, A.F.; Vincelette, N.D.; Lam, N.B.; Aldrich, A.L.; Al Ali, N.H.; Padron, E.; et al. Oxidized Mitochondrial DNA Released after Inflammasome Activation Is a Disease Biomarker for Myelodysplastic Syndromes. Blood Adv. 2021, 5, 2216–2228.
More