Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Clelia De-la-Peña and Version 2 by Jessie Wu.
Epigenetic regulation has the potential to revolutionize plant breeding and improve crop yields by regulating gene expression in plants. DNA methylation and histone modifications are key epigenetic modifications that can impact plant development, stress responses, productivity, and yields.
- DNA methylation
- histone posttranslational modifications
- sRNAs
- heterosis
- plant vigor
1. Epigenetic Regulation
1.1. DNA Methylation
DNA methylation is a well-studied epigenetic mechanism that regulates gene expression by altering chromatin conformation. When a specific DNA region is enriched with a methylation mark, the chromatin adopts a closed configuration, leading to gene silencing. Conversely, in the absence or reduced presence of methylation, the chromatin assumes an open configuration, promoting gene expression. This mechanism involves the addition of a methyl group to the fifth carbon of cytosine and is catalyzed by enzymes known as methyltransferases (METs). In plants, DNA methylation occurs in three different sequence contexts: CG, CHG, and CHH, where H represents any nucleoside except guanine [1][25]. Maintenance and de novo DNA methylation take place during DNA replication. CG and CHG methylation patterns are maintained by DNA METHYLTRANSFERASE 1 (MET1) and CHROMOMETHYLASE 3 (CMT3), respectively, while asymmetric CHH methylation is established through de novo methylation catalyzed by CMT3 and DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) [2][3][26,27]. Additionally, DRM3 plays a crucial role in initiating de novo cytosine methylation in all sequence contexts through a process called RNA-directed DNA methylation (RdDM) [4][28]. In the RdDM pathway, a complementary sequence within a heterochromatic region is transcribed by POL IV, leading to the synthesis of small RNAs (ssRNAs). These ssRNAs are then converted into double-stranded RNAs (dsRNAs) through the action of RNA-directed RNA polymerase 2 (RDR2). The dsRNAs are subsequently sliced into 24-nucleotide fragments, and one strand from each 24-nucleotide double-stranded small RNA is loaded into ARGONAUTE (AGO), forming an RNA–protein complex that recognizes and binds to complementary target sequences. This interaction recruits DRM3, which methylates the neighboring DNA [5][29]. The CHH context is dependent on 24-nucleotide small interfering (si)RNAs to guide the methyltransferases, or on a second pathway involving DEFICIENT IN DNA METHYLATION 1 (DDMI) and CMT2 [6][30].
1.2. Histone Modifications
Eukaryotic DNA is packaged into chromatin, which is composed of nucleosomes consisting of histones H2A, H2B, H3, and H4 [7][8][13,31]. The N-terminal tails of histones are subject to different posttranslational modifications, including acetylation, methylation, ubiquitination, phosphorylation, biotinylation [9][32], adenosine diphosphate (ADP)-ribosylation [10][33], crotonylation [11][34], and sumoylation [12][35], among others [7][8][13][14][13,31,36,37], which regulate the chromatin structure and accessibility to DNA. In plants, histone acetylation usually occurs at lysine residues of histones H3 and H4 and is associated with transcriptional gene activation [15][16][16,18]. Histone methylation, such as H3K9me2 and H3K27m3, is linked to gene repression [7][17][13,38], while the H3K36me2 and H3K36me3 marks are associated with gene transcriptional activation [7][18][19][13,39,40]. Overall, histone posttranslational modifications contribute to the establishment of a histone code that regulates gene expression and the chromatin structure [9][32].
1.3. Small RNAs
Gene expression and epigenetic control are regulated by small RNAs, including microRNAs (miRNAs), small interfering RNAs (siRNAs), and trans-acting small interfering RNAs (ta-siRNAs). MiRNAs are short regulatory RNAs, approximately 19–24 nucleotides in length, that negatively regulate gene expression. MiRNAs are synthesized by DNA-dependent RNA Pol II from MIR genes and are derived from a hairpin or stem–loop precursor [20][21][22][41,42,43]. The primary miRNA (pri-miRNA) is first cleaved by the RNase III DICER-Like1 (DCL1) to create the intermediate precursor pre-miRNA [23][44]. DCL1 then cleaves the pre-miRNA to form the mature miRNA duplex along with the dsRNA-binding protein HYPONASTIC LEAVES1 (HYL1) in the nucleus [20][41]. The nuclear methyltransferase HUA ENHENCER1 (HEN1) attaches a methyl group to the 2′ OH of the mature duplex miRNA’s 3′ last nucleotide. The Arabidopsis EXPORTIN5 ortholog HASTY transports the miRNA from the nucleus to the cytoplasm and the methyl groups are removed in the cytoplasm. A helicase unwinds the double-stranded mature miRNA to produce a single-stranded mature miRNA that is recognized by ARGONAUTE 1 (AGO1) [20][41]. AGO1 recruits the entire RNA-induced silencing complex (RISC) that recognizes the mRNA targets that the mature miRNA regulates [20][24][41,45]. AGO1 works with AMP1 (ALTERED MERISTEM PROGRAM1) to suppress the translation of target mRNAs in the endoplasmic reticulum [25][26][46,47]. The biogenesis of miRNAs can produce two types of miRNAs: those that perfectly complement their mRNA targets and those that have mismatches with their targets. MiRNAs with perfect matches to their target mRNAs induce mRNA cleavage, while miRNAs containing mismatches suppress translation by binding stably to the mRNA targets [21][22][27][42,43,48].
2. Factors Affecting Epigenetic Mechanisms and, Therefore, Productivity
Plants are constantly exposed to various environmental stresses, such as nutrient deficiency, drought, heat, salinity, and soil contamination with heavy metals. These stressors can have detrimental effects on plant growth, biomass production, and overall yields (Figure 1). In order to mitigate losses in agriculture, it is imperative to develop stress-resistant cultivars that can better withstand these challenging conditions. A key aspect in achieving this goal is gaining a deeper understanding of plant stress responses and their regulation, specifically focusing on the chromatin states and histone modification that govern gene expression. By unraveling these epigenetic mechanisms, researchers can uncover novel targets for crop enhancement, leading to the creation of more productive and resilient plants capable of adapting to changing environmental conditions. This research is of the utmost importance for improving agronomic traits and enhancing productivity, thereby ensuring food security in the face of evolving climate change and other environmental pressures [28][29][1,74].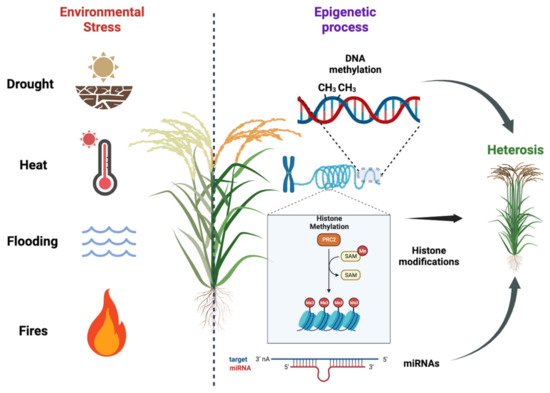
Figure 1. Plant responses to environmental stresses and the importance of epigenetic regulation on hybrid vigor. Plants face various environmental stresses, impacting growth and yields. Developing stress-resistant cultivars is crucial for agriculture. Understanding plant stress responses and epigenetic regulation, including DNA methylation, histone modification, and miRNA regulation, helps identify targets for crop enhancement. ResWearchers used BioRender (BioRender.com) to create this scientific illustration.
2.1. Heat Stress
Temperature is a crucial environmental factor affecting plant growth, biomass, and yields. Temperature changes, both heat and cold, pose a significant challenge to agriculture. Heat stress, in particular, can lead to morphological, physiological, and biochemical changes in plants, including growth retardation, leaf etiolation, and even death [30][75]. Heat stress induces signaling cascades and triggers the expressions of specific genes [31][76] and heat-shock proteins (HSPs) [32][77]. Studies have shown that different plant genotypes exhibit varying degrees of heat tolerance. The responses of plants to temperature stress involve epigenetic mechanisms, specifically histone posttranscriptional modifications [33][34][35][23,78,79]. To investigate the impact of heat stress on methylation patterns, researchers have examined methylation levels and changes in cytosine methylation patterns in seedlings of heat-sensitive and heat-tolerant genotypes. The findings revealed that the methylation levels differed between the heat-tolerant and heat-sensitive phenotypes under normal conditions [36][80]. Upon exposure to heat treatment, methylation increased to a greater extent in the heat-sensitive genotype compared to the heat-tolerant genotype. Interestingly, DNA demethylation events were more prevalent in the heat-tolerant genotype, whereas DNA methylation occurred more frequently in the heat-sensitive genotype. This suggests that changes in DNA methylation patterns are associated with the heat-stress response and adaption in B. napus L. [37][81] (Table 12). Intriguingly, through the use of an MSAP assay, a polymorphic demethylated fragment known as M7 (digested with EcoRI/MspI) was identified that was found to be linked to a calcium-transporting ATPase gene. This gene plays a crucial role in facilitating the direct transport of calcium ions [38][82]. The primary calcium-transporting ATPase present in the plasma membrane and endoplasmic reticulum of plant cells utilizes ATP hydrolysis to transport calcium ions. Thus, the alteration of the Ca2+ concentration in the cytoplasm due to stress could serve as a primary transduction mechanism, influencing gene expression and biochemical events to enable plant cells to adapt to environmental stresses, including heat stress [39][83]. The role of histone acetylation, mediated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), has been highlighted in the response to heat stress [16][40][15,18] (Table 12). Heat stress triggers thermomorphogenesis in Arabidopsis, characterized by elongated growth and early flowering, enhancing the cooling capacity of the plant [40][41][15,84]. HDACs, such as HDA9, play a crucial role in thermomorphogenesis by promoting the expressions of genes involved in this response. For instance, HDA9 interacts with PRW (POWERDRESS) to increase the deacetylation of H3K9 at specific gene loci, such as PHYTOCHROME INTERACTING FACTOR4 (PIF4) and YUCCA8 (YUC8), which are essential for thermomorphogenesis [42][85] (Table 12). HDA9 activity is also required for YUC8 expression via the promotion of the eviction of the histone variant H2A.Z from YUC8 nucleosomes, leading to histone deacetylation at the transcriptional start site and gene body of YUC8 and allowing its transcriptional activation by PIF4 [43][86]. These findings suggest that histone acetylation and deacetylation could be a valuable strategy for enhancing crop yields under heat-stress conditions, thereby potentially impacting heterosis. In Arabidopsis, the activity of HDA15 has been shown to act as a repressor of the response induced by warm temperatures [44][87], while HDA9 and HDA19 appear to participate indirectly in the response to the same stimulus [45][88]. At 27 °C, hda15 mutant seedlings showed elongated hypocotyls compared to Col-0 plants, while the hypocotyls were shorter in hda9 and hda19 mutant seedlings. Furthermore, warm-temperature marker genes, such as HSP20, IAA3, IAA19, IAA29, YUC8, SAUR28, and TCH3, were upregulated in the hda15 mutant compared to the hda9 and hda19 mutants and Col-0 plants. In addition, HSP20, IAA19, and IAA29 genes showed increased levels of H3K14ac in their promoter and 5′ regions. At 20 °C, the hda15 mutant also showed the upregulation of warm-temperature marker genes, such as YUCCA8, IAA19, IAA29, TCH3, ATHB2, and XTR7. These results suggest that HDA15 can repress warm-temperature marker genes during normal growth and dissociate from its targets to induce their expressions under elevated-temperature stimuli [45][88].2.2. Drought Stress
Water deficiency is a major challenge in agriculture, and plants have been found to respond to this stress through epigenetic modifications, including histone acetylation [15][46][16,89]. The dynamic activity of HATs/HDAs regulates the response to drought stress in important crops such as rice, wheat, and cotton [37][47][48][81,90,91]. In Arabidopsis, H3K9ac has been shown to positively regulate the expressions of drought-response genes [49][92]. The dynamic activity of HATs and HDAs also regulates the ABA biosynthesis pathway, which is the most important signaling pathway for drought stress in plants and is found in various plant species [15][16][16,18]. Epigenetic associations with heterosis in response to drought stress have also been observed [50][51][10,93]. A study conducted on poplar (Populus euramericana) examined six hybrid genotypes (P. deltoides × P. nigra) subjected to water-deficit conditions. The results revealed a correlation between the morphological traits related to productivity and epigenetic modifiers under drought stress. In the hybrid genotype Populus deltoides × P. nigra, the hypomethylation of DNA was found to be associated with drought stress, while there was a significant increase in histone acetylation, indicating rapid gene expression potentially linked to heat-shock proteins (HSPs) [51][93] (Table 12). These findings highlight the potential role of epigenetic mechanisms in mediating heterosis and enhancing drought-tolerance traits in plants. Various studies have shown a positive correlation between increased HAT expression and drought tolerance in plants [15][16][16,18]. In Brassica rapa, the expressions of nine HAT genes, including BraHAC1, BraHAC2, and BraHAC3, increased significantly after two and/or four days of drought treatment [52][94]. Similarly, in Brachypodium distachyon and Oryza sativa, the expressions of five HATs (BdHAG1, BdHAG3, BdHAC1, BdHAC4, and BdHAF1) and nine HATs (OsHAG702//703, OsHAD704/705/706/711/712/713, and OsHAM701), respectively, were induced after drought treatment [47][53][90,95] (Table 1). Analysis of the promoter region of some of these HAT genes, such as OsHAG702, OsHDA705/706/713, and OsSRT702, showed the existence of drought-responsive elements, like the MBS cis-element (MYB-binding site involved in drought inducibility), indicating the participation of specific transcription factors for gene activation [47][90]. In wheat, the genes TaHAG2, TaHAG3, and TaHAC2, and particularly TaHAG2, showed significantly higher expressions in the drought-resistant variety BL207 compared to its less-resistant parents, BN64 and ZM16. This indicates the potential involvement of these genes in the drought response of wheat [48][91]. In Arabidopsis, drought stress triggered an increase in the H3K9ac levels within the promoter regions of 14 drought-response genes, suggesting a crucial role for H3K9ac in the transcriptional activation of these genes under water-deficit conditions [49][92]. This mechanism suggests the formation of tertiary protein complexes that enhance gene expression [54][96]. HDAs generally appear to negatively regulate the expressions of drought-responsive genes. For instance, the HDA9 mutation in Arabidopsis resulted in the upregulation of 47 water-deprivation-response genes and the downregulation of 13 genes compared to wild-type plants. The promoter region of 14 randomly selected upregulated genes in the hda9 mutant showed increased levels of H3K9ac (>2-fold), indicating that the increased expressions of these genes are due to a decrease in deacetylase activity [49][92]. Similarly, plants that silenced AtHDA6 and AtHDA19 exhibited a hypersensitive phenotype to ABA, resulting in the decreased expressions of ABA-responsive genes (KAT1, KAT2, ABI1, ABI2, RD29A, RD29B, and DREB2A) when treated with ABA [44][87]. AtHD2C has been implicated in the response to ABA. Transgenic plants overexpressing AtHD2C exhibited insensitivity to ABA and demonstrated enhanced drought tolerance compared to wild-type plants. Furthermore, the expression of AtHD2C was repressed by ABA [55][97], and AtHD2C can physically interact with HDA6 and function in association to regulate the expressions of ABA-responsive genes [56][98]. Recent studies indicate that AtHDA15, through the transcription factor MYB96, can regulate gene responses mediated by ABA signaling [57][58][99,100]. The biochemical and molecular mechanisms by which HDA6, HDA9, and HDA15 act to regulate responsive genes for ABA signaling have been described in detail in previous studies [15][57][59][60][16,99,101,102]. In soybean (Glycine max), the expressions of the nine GmHDACs (GmHDA6, GmHDA8, GmHDA13, GmHDA14, GmHDA16, GmSRT2, GmSRT4, GmHDT2, and GmHDT4) were found to decrease after drought treatment [61][103]. Similarly, in rice, the expression of OsHDA703/710 was significantly decreased after drought treatment [47][90]. In wheat, the drought-resistant variety BL207 showed a downregulation of the expressions of TaHDA2, TaHDA18, and TaHDT2 [48][91]. However, in some cases, an increase in HDAC expression may occur, potentially inhibiting the function of the transcriptional repressors of drought-stress-response genes. For example, in rice, increases in the expressions of OsHAG702/703, OsHAM701, OsHDA704/705/706/711/712/713, OsHDT701, and OsSRT702 were observed after drought treatment [47][90], and in Hibiscus cannabinus L., five HcHDA genes (HcHDA2, HcHDA6, HcHDA9, HcHDA19, and HcSRT2) were strongly expressed under PEG treatment [62][104]. The use of epigenetic mechanisms offers promising strategies for enhancing drought tolerance in plants. Modulating the expression or repression of HDAC has shown significant impacts on drought tolerance in different plant species. For instance, in tobacco, introducing the histone deacetylase 84 KHDA903 from poplar (Populus alba × Populus glandulosa) resulted in the overexpressions of drought-responsive genes (NtDREB4, NtDREB3, and NtLEA), leading to improved drought tolerance [63][105]. In cotton, overexpressing the histone deacetylase GhHDT4D, a member of the HD2 subfamily, enhanced drought tolerance by reducing the H3K9ac levels in the promoter region of GhWRKY33, a negative regulator of cotton’s response to drought, and suppressing its expression [37][81]. In Arabidopsis, AtHD2C and HDA6 were found to decrease the expressions of ABA-responsive genes by reducing histone H3K9/K14 acetylation and increasing H3K9me2 [56][98]. Conversely, H3K4me3 appears to play an important role in the response to drought stress in Arabidopsis, as the 5′ ends of most ABA and dehydration-inducible genes exhibited broader H3K4me3 distribution profiles [64][106]. These findings suggest that alterations in HDAC expression and histone modifications are involved in the plant response to drought stress and hold potential for early stress detection. Additionally, manipulating the transcriptional activation or repression of HDAC can offer promising avenues for improving drought tolerance across different plant species.Table 1.
Plant responses to stress and epigenetic processes in different plant species.
| Plant Response | Epigenetic Process | Plant Species | Function | Reference |
|---|---|---|---|---|
| Heat stress | Histone modification | Arabidopsis thaliana | HDA9 interacts with the PWR protein and increases H3K9 deacetylation at the +1 nucleosomes of PHYTOCHROME INTERACTING FACTOR 4 (PIF4) and YUCCA8 (YUC8), essential genes regulating thermomorphogenesis. | [42][85] |
| HDA9 promotes the eviction of the histone variant H2A.Z from the YUC8 nucleosome and enables its transcriptional activation by PIF4, mediating the thermomorphogenic response. | [43][86] | |||
| HDA15 acts as a repressor of warm-temperature marker genes (YUCCA8, IAA19, IAA29, TCH3, ATHB2, and XTR7) under normal conditions but dissociates from its targets under elevated-temperature stimuli, inducing their expressions. | [45][88] | |||
| DNA methylation | Brassica napus | Exhibits more DNA demethylation events in heat-tolerant genotypes, which are associated with heat-stress response and adaptation. | [36][80] | |
| Drought stress | DNA methylation/histone modification | Populus deltoides × P. nigra | Shows genotypic variation in DNA hypomethylation that correlates with morphological traits related to productivity under drought stress. Histone acetylation induces rapid gene expression associated with heat-shock proteins (HSPs) under drought-stress conditions. | [51][93] |
| Histone modification | Arabidopsis thaliana | HDA9 negatively regulates plant sensitivity to drought stresses through increased H3K9ac levels in the promoter region of 14 drought-response genes under water-deficit conditions. | [49][92] | |
| AtHD2C physically interacts with HDA6 and regulates the expressions of ABA-responsive genes in association. | [55][56][97,98] | |||
| Brachypodium distachyon | Exhibits increased expressions of five HAT genes (BdHAG1, BdHAG3, BdHAC1, BdHAC4, BdHAF1) under drought treatment, playing a role in drought-stress response and adaptation. | [53][95] | ||
| Brassica rapa | Demonstrates a significant increase in the expressions of nine HAT genes (BraHAC1, BraHAC2, BraHAC3, BraHAC4, BraHAC7, BraHAG2, BraHAG5, BraHAG7, and BraHAF1) after drought treatment, contributing to drought-stress response and adaptation. | [52][94] | ||
| Gossypium hirsutum | Enhanced drought tolerance by reducing H3K9ac levels in the promoter region of GhWRKY33, a negative regulator of drought response, through the action of GhHDT4D, a member of the histone deacetylase HD2 subfamily. | [37][81] | ||
| Dendrobium officinale | Induces the expressions of DoHDA10 and DoHDT4 genes in roots, stems, and leaves under drought-stress conditions. | [65][107] | ||
| Oryza sativa | Triggers the expressions of nine HAT (OsHAG702//703, OsHAD704/705/706/711/712/713, and OsHAM701) genes under drought conditions. Some HAT genes contain drought-sensitive elements, such as the MBS cis element, in their promoter regions. | [47][90] | ||
| Triticum aestivum | Demonstrates the downregulation of five HDA genes and a significant increase in TaHAC2 expression in the drought-resistant variety BL207 under drought-stress conditions. | [48][91] |