Spermatocytic tumor (ST) is a very rare disease, accounting for approximately 1% of testicular cancers. Previously classified as spermatocytic seminoma, it is currently classified within the non-germ neoplasia in-situ-derived tumors and has different clinical-pathologic features when compared with other forms of germ cell tumors (GCTs). A web-based search of MEDLINE/PubMed library data was performed in order to identify pertinent articles. In the vast majority of cases, STs are diagnosed at stage I and carry a very good prognosis. The treatment of choice is orchiectomy alone. Nevertheless, there are two rare variants of STs having very aggressive behavior, namely anaplastic ST and ST with sarcomatous transformation, that are resistant to systemic treatments and their prognosis is very poor.
1. Introduction
Testicular tumors are classified as germ-cell-origin and sex-cord-stroma tumors
[1]. Germ cell tumors (GCTs) include seminoma and non-seminoma histologies; the latter encompasses teratoma (postpubertal type), embryonal carcinoma (EC), choriocarcinoma, yolk sac tumor (YST) and a mixture of these components, including seminoma. Pure histology is a rare event and frequently, non-seminoma are composed of multiple histologies, including seminoma components (so-called mixed germ cell tumors).
Spermatocytic tumor (ST) is a rare GCT derived from postpubertal-type germ cells and was previously called spermatocytic seminoma due to a false belief in its origin from germ cell neoplasia in situ
[2][4].
Differently from other GCTs, ST is not known to occur in extratesticular sites, and this tumor has an indolent course
[3][4][5][5,6,7]. Most STs of the testis are considered benign tumors. As STs are extremely rare, there are limited data on the optimal management of patients having a localized or a metastatic disease.
2. Epidemiology
ST is a rare testis tumor, accounting for about 1% of all testicular tumors
[6][9]. Although its incidence has been reported at around 52–56 years of age, it has never been considered as a tumor of the elderly population
[4][6][7][8][9][6,9,10,11,12]. Nearly 70% of patients are over 40 years of age, and it is virtually absent in children, adolescents and young adults, although the youngest patient described in the literature was 19 years old
[6][9].
In an Australian series among 9658 cases of primary testicular tumors in the period 1982–2002, only 58 cases of STs were reported, and the Australian age-standardized incidence rate for spermatocytic seminoma was 0.4 cases per million. The mean number of cases diagnosed per year was 2.8, ranging from 0 to 7. In particular, the standardized incidence was 0.3 cases per million for men younger than 55 and 0.8 per million for men 55 years or older
[6][9]. The mean patient age at diagnosis was 53.5 years, 50% of men were diagnosed at 54 years or younger, and approximately 25% of patients were younger than 40 years
[6][9]. This cohort of patients reported not only the youngest patients with ST (19 years old), but also the oldest patient reported in literature, who was 92 years old
[6][9]. In addition, the Australian series detected no statistical increase in the incidence of STs in the previous 20 years, but the ability of the authors to identify trends was limited by the small number of cases available
[6][9].
3. Pathophysiology
Spermatocytic tumor was considered closely related to seminoma and classified as subtype of this disease from the time of its first description in 1946 by Masson, who named this entity as “le seminome spermatocitaire”
[10][16].
The reason for the WHO classification change
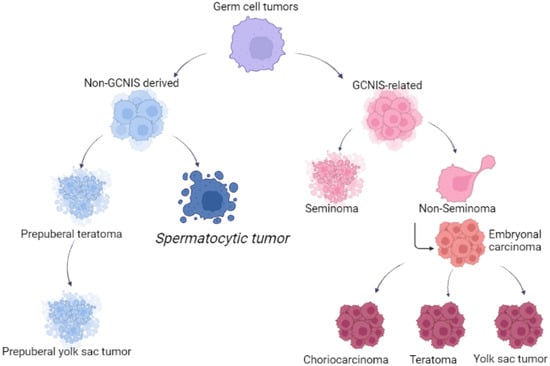
[2][4] was due to several important differences. First, STs have a different origin compared with other GCTs: they derive from more mature germ cells (likely premeiotic germ cells at a transition stage between spermatogonia and spermatocytes), in contrast to postpubertal-type germ cell tumors such as seminoma, which are thought to originate from primordial germ cells/gonocytes. Increased recognition of this distinction led to a revision in the nomenclature of these tumors from spermatocytic seminoma to ST, particularly to avoid overlapping terminology with GCNIS-derived seminoma. Second, unlike seminoma, STs do not exhibit gains of the short arm of chromosome 12, which is a frequent finding in (GCNIS-derived) postpuberal type germ cell tumors
[11][12][17,18]. Third, STs show a unique amplification of chromosome 9, corresponding to the DMRT1 gene, not reported in all GCNIS-related tumors, but also in prepuberal teratoma and prepuberal YST
[13][19]. The DMRT1 gene, located at the end of the 9th chromosome, is found in a cluster with two other members of the gene family (DMRT2, DMRT3), having in common a zinc finger-like DNA-binding motif (DM domain). The DMRT1 protein is located in the spermatogonia and in the spermatocytes, specifically in the nucleus, and its role is critical during embryogenesis and male germ cell maturation.
Figure 1 reported the different origins of GCTs and STs.
Figure 1. Different origin of germ cell tumors and spermatocytic tumors.
4. Histopathology
ST is a germ cell tumor, derived from postpubertal-type germ cells; tumor cells have a close resemblance to spermatogonia or early primary spermatocytes. Macroscopically, STs have sharp borders, the cut surface is usually grey-white and glistening, mucoid material may be present and foci of necrosis are hardly detected. STs usually display a multinodular or diffuse pattern; very rarely, they can present with a pseudoglandular, cystic, trabecular or nested pattern. The tumors are arranged in diffuse sheets and usually lack fibrous septa and lymphoid infiltrates.
Neoplastic elements are heterogeneous. There are three types of cells: (a) small cells, 6–8 microns, with scant cytoplasm; (b) intermediate cells, 15–20 microns, with round nuclei and chromatin ranging from granular to spireme-type; and (c) giant cells, 50–100 microns, with multiple nuclei.
Unlike GCTs, there is no germ cell neoplasia in situ (GCNIS), and no significant inflammatory infiltrate, granulomas, fibrovascular septa or cytoplasmic glycogen can be observed; these features may prove useful in the differential diagnosis with classic seminoma.
In the past, STs with a clear prevalence of nucleolated intermediate cells were termed as “anaplastic”; this description is currently not encouraged, as “anaplastic STs” seem to share the same biological behaviour as classical STs. Some authors have reported anaplastic ST in some tumors having parameters such as increased mitotic count (>30/10 HPF), the presence of prominent nucleoli, vesicular nuclei, bizarre giant forms, areas of necrosis and abnormal mitotic figures [14], but they in their reported series, did not find any cases that fit the given criteria [5].
ST undergoing sarcomatous transformation, which occurs in approximately 6% of cases, is a rare but well-documented event [8]. The sarcomatous component can be either intermixed with classic areas of ST or may be a separate nodule lying adjacent to the classic ST. The sarcomatous component is usually an undifferentiated spindle cell sarcoma, but sarcoma with rhabdomyosarcoma and chondroid differentiation have also been reported [5]. As documented in the literature, ST with a sarcomatous component has the same age incidence as pure ST [15].
Different origin of germ cell tumors and spermatocytic tumors.
4. Histopathology
ST is a germ cell tumor, derived from postpubertal-type germ cells; tumor cells have a close resemblance to spermatogonia or early primary spermatocytes. Macroscopically, STs have sharp borders, the cut surface is usually grey-white and glistening, mucoid material may be present and foci of necrosis are hardly detected. STs usually display a multinodular or diffuse pattern; very rarely, they can present with a pseudoglandular, cystic, trabecular or nested pattern. The tumors are arranged in diffuse sheets and usually lack fibrous septa and lymphoid infiltrates.
Neoplastic elements are heterogeneous. There are three types of cells: (a) small cells, 6–8 microns, with scant cytoplasm; (b) intermediate cells, 15–20 microns, with round nuclei and chromatin ranging from granular to spireme-type; and (c) giant cells, 50–100 microns, with multiple nuclei.
Unlike GCTs, there is no germ cell neoplasia in situ (GCNIS), and no significant inflammatory infiltrate, granulomas, fibrovascular septa or cytoplasmic glycogen can be observed; these features may prove useful in the differential diagnosis with classic seminoma.
In the past, STs with a clear prevalence of nucleolated intermediate cells were termed as “anaplastic”; this description is currently not encouraged, as “anaplastic STs” seem to share the same biological behaviour as classical STs. Some authors have reported anaplastic ST in some tumors having parameters such as increased mitotic count (>30/10 HPF), the presence of prominent nucleoli, vesicular nuclei, bizarre giant forms, areas of necrosis and abnormal mitotic figures [20], but they in their reported series, did not find any cases that fit the given criteria [7].
ST undergoing sarcomatous transformation, which occurs in approximately 6% of cases, is a rare but well-documented event [11]. The sarcomatous component can be either intermixed with classic areas of ST or may be a separate nodule lying adjacent to the classic ST. The sarcomatous component is usually an undifferentiated spindle cell sarcoma, but sarcoma with rhabdomyosarcoma and chondroid differentiation have also been reported [7]. As documented in the literature, ST with a sarcomatous component has the same age incidence as pure ST [21].
The differential diagnosis of ST should be made with seminoma, particularly with microcystic patterns [22], embryonal carcinoma [23] and malignant lymphoma [24,25]. In each case, both morphological and immune phenotypical features are to be considered.
Seminoma is characterized by homogeneous neoplastic cells; fibrovascular septa are usually present, as well as a chronic inflammatory infiltrate, possibly comprising granulomas; and GCNIS foci are easily spotted. As with ST, seminoma is CD117/ckit+; however, typical seminoma is reactive with GCT markers, such as OCT3/4, PLAP and D2-40. In contrast, increased immunohistochemical reactivity for FGFR3, HRAS and DMRT1 has been shown in ST [26].
Embryonal carcinoma has markedly pleomorphic tumor cells, and a variety of architectural patterns (glandular, cystic, papillary) are rarely observed in ST. Despite the “anaplastic” nature of this uncommon ST variant, they still retain the round-nuclei characteristic of this tumor, whereas those of embryonal carcinoma are more irregular in almost all instances. The typical staining for CD30/BerH2 and OCT3/4 allows the diagnosis of embryonal carcinoma [23].
5. Clinical Features
STs usually present with a unilateral mass. Bilateral involvement has been reported, even though is more often documented in ST compared with other germ cell tumors [11,25,28,29]. One of the large series published showed bilateral STs in 5% of testicular tumor; one patient (1%) had synchronous bilateral mass in undescended testes at age 67, and 2 patients (3%) had metachronous bilateral testicular masses at intervals of 10 and 16 years, respectively [11].
The differential diagnosis of ST should be made with seminoma, particularly with microcystic patterns [16], embryonal carcinoma [17] and malignant lymphoma [18][19]. In each case, both morphological and immune phenotypical features are to be considered. Variable tumor size has been recorded, ranging from 2 to 20 cm with an average of 7–8 cm [7,30]. Nevertheless, though ST patients are more likely to have larger tumors on presentation with respect to seminoma, they are less likely to have <pT2 stage, and only 16.4% of STs are smaller than 3 cm, compared with 27.6% in the seminoma group [12]. In this cohort, patients with ST were nearly twice as likely as classic seminoma patients to have tumors larger than 3 cm. While tumor size is generally greater in ST, these data do not translate into greater locally advanced or metastatic disease. Indeed, patients with ST are also less likely to have regional nodal involvement (clinical stage II) or distant metastases on presentation, compared with seminoma [12].
Seminoma is characterized by homogeneous neoplastic cells; fibrovascular septa are usually present, as well as a chronic inflammatory infiltrate, possibly comprising granulomas; and GCNIS foci are easily spotted. As with ST, seminoma is CD117/ckit+; however, typical seminoma is reactive with GCT markers, such as OCT3/4, PLAP and D2-40. In contrast, increased immunohistochemical reactivity for FGFR3, HRAS and DMRT1 has been shown in ST [20]. Due to the indolent nature of the tumor, there is often some appreciable interval from the time of initial recognition of a lesion by the patients to their seeking medical attention, and so most patients developed large testicular mass before diagnosis [11].
Embryonal carcinoma has markedly pleomorphic tumor cells, and a variety of architectural patterns (glandular, cystic, papillary) are rarely observed in ST. Despite the “anaplastic” nature of this uncommon ST variant, they still retain the round-nuclei characteristic of this tumor, whereas those of embryonal carcinoma are more irregular in almost all instances. The typical staining for CD30/BerH2 and OCT3/4 allows the diagnosis of embryonal carcinoma [17].
The association of ST with cryptorchidism is extremely rare, as only two cases are reported in the literature so far [11,33]. Cryptorchidism is a well-known risk factor for testicular cancer [34], but does not appear to be a risk for ST. No reported case had an association with either changes in testosterone or estrogen levels, or clinical signs of gynecomastia [31].
5. Clinical Features
While serum tumor markers such as gonadotropin (beta-HCG), alpha-fetoprotein (alfaFP), and lactate dehydrogenase (LDH) are expressed in about 50–60% of GCT patients [35], they are reported as increased in less than 1% of ST patients [31]. Sonography details were infrequently reported, such as calcification in one patient and heterogeneous appearance in another [31].
STs usually present with a unilateral mass. Bilateral involvement has been reported, even though is more often documented in ST compared with other germ cell tumors [8][19][21][22]. One of the large series published showed bilateral STs in 5% of testicular tumor; one patient (1%) had synchronous bilateral mass in undescended testes at age 67, and 2 patients (3%) had metachronous bilateral testicular masses at intervals of 10 and 16 years, respectively [8]. STs never arise in extragonadal sites such as the anterior mediastinum, retroperitoneum, pineal gland and sacrococcygeal area, and there is no ovarian homologue, as is the case for seminoma (so-called dysgerminoma), although one case has been described in a dysgenetic gonad of a phenotypic female who had an associated gonadoblastoma [36].
Variable tumor size has been recorded, ranging from 2 to 20 cm with an average of 7–8 cm [5][23]. Nevertheless, though ST patients are more likely to have larger tumors on presentation with respect to seminoma, they are less likely to have <pT2 stage, and only 16.4% of STs are smaller than 3 cm, compared with 27.6% in the seminoma group [9]. In this cohort, patients with ST were nearly twice as likely as classic seminoma patients to have tumors larger than 3 cm. While tumor size is generally greater in ST, these data do not translate into greater locally advanced or metastatic disease. Indeed, patients with ST are also less likely to have regional nodal involvement (clinical stage II) or distant metastases on presentation, compared with seminoma [9]. 6. Treatment
Due to the indolent nature of the tumor, there is often some appreciable interval from the time of initial recognition of a lesion by the patients to their seeking medical attention, and so most patients developed large testicular mass before diagnosis [8]. Surgical orchiectomy is the standard treatment for STs, as testis-sparing techniques are limited by the difficulties for the pathologist in differentiating ST from seminoma GCT in a frozen section [37].
As STs rarely metastasize, surgery alone is the standard of care. In the past, adjuvant radiotherapy was performed, but it has gone into disrepute in recent studies [7,38].
The association of ST with cryptorchidism is extremely rare, as only two cases are reported in the literature so far [8][24]. Cryptorchidism is a well-known risk factor for testicular cancer [25], but does not appear to be a risk for ST. No reported case had an association with either changes in testosterone or estrogen levels, or clinical signs of gynecomastia [26]. The US study also compared survival outcomes for men with ST relative to classic seminoma. After adjusting for differences in baseline demographics, such as age and comorbidity, as well as stage distribution, they observed no differences in overall survival between the two groups [12]. The favorable survival outcomes for ST, despite a reduction in the use of adjuvant therapy after orchiectomy over time, is encouraging.
While serum tumor markers such as gonadotropin (beta-HCG), alpha-fetoprotein (alfaFP), and lactate dehydrogenase (LDH) are expressed in about 50–60% of GCT patients [27], they are reported as increased in less than 1% of ST patients [26]. Sonography details were infrequently reported, such as calcification in one patient and heterogeneous appearance in another [26]. On the other hand, STs with sarcoma transformation or anaplastic subtypes showed metastatic disease in 45% and 29% of cases, respectively; compared to ST, patients having these histological variants were more likely to have metastatic disease. These subtypes are highly resistant to cytotoxic chemotherapy and they represent a very aggressive disease: patients have a poor prognosis, with a median survival of 5 months [31,41]. Despite the high risk of metastases, the guideline of treatment for STs with sarcomatous transformation has yet to be established, albeit adjuvant chemotherapy seems to be a possible choice. In the literature, five of these patients received platinum-based chemotherapy, and all responded poorly and died shortly (within 3–14 months) after diagnosis [12]. However, only one case of metastatic ST responded well to the VIP combination chemotherapy after radical orchiectomy [42].
STs never arise in extragonadal sites such as the anterior mediastinum, retroperitoneum, pineal gland and sacrococcygeal area, and there is no ovarian homologue, as is the case for seminoma (so-called dysgerminoma), although one case has been described in a dysgenetic gonad of a phenotypic female who had an associated gonadoblastoma [28].
6. Treatment
Surgical orchiectomy is the standard treatment for STs, as testis-sparing techniques are limited by the difficulties for the pathologist in differentiating ST from seminoma GCT in a frozen section [29].
As STs rarely metastasize, surgery alone is the standard of care. In the past, adjuvant radiotherapy was performed, but it has gone into disrepute in recent studies [5][30].
The US study also compared survival outcomes for men with ST relative to classic seminoma. After adjusting for differences in baseline demographics, such as age and comorbidity, as well as stage distribution, they observed no differences in overall survival between the two groups [9]. The favorable survival outcomes for ST, despite a reduction in the use of adjuvant therapy after orchiectomy over time, is encouraging.
On the other hand, STs with sarcoma transformation or anaplastic subtypes showed metastatic disease in 45% and 29% of cases, respectively; compared to ST, patients having these histological variants were more likely to have metastatic disease. These subtypes are highly resistant to cytotoxic chemotherapy and they represent a very aggressive disease: patients have a poor prognosis, with a median survival of 5 months [26][31]. Despite the high risk of metastases, the guideline of treatment for STs with sarcomatous transformation has yet to be established, albeit adjuvant chemotherapy seems to be a possible choice. In the literature, five of these patients received platinum-based chemotherapy, and all responded poorly and died shortly (within 3–14 months) after diagnosis [9]. However, only one case of metastatic ST responded well to the VIP combination chemotherapy after radical orchiectomy [32].