Mitochondrion, known as the “powerhouse” of the cell, regulates ion homeostasis, redox state, cell proliferation and differentiation, and lipid synthesis. The inner mitochondrial membrane (IMM) controls mitochondrial metabolism and function. It possesses high levels of proteins that account for ~70% of the membrane mass and are involved in the electron transport chain, oxidative phosphorylation, energy transfer, and ion transport, among others. The mitochondrial matrix volume plays a crucial role in IMM remodeling. Several ion transport mechanisms, particularly K+ and Ca2+, regulate matrix volume. Small increases in matrix volume through IMM alterations can activate mitochondrial respiration, whereas excessive swelling can impair the IMM topology and initiates mitochondria-mediated cell death. The opening of mitochondrial permeability transition pores, the well-characterized phenomenon with unknown molecular identity, in low- and high-conductance modes are involved in physiological and pathological increases of matrix volume.
- mitochondria
- ions
- permeability transition pore
- heart
- ischemia-reperfusion
1. Introduction
2. Mitochondrial Membrane Structure and Organization
2.1. Mitochondrial Membrane Composition
The complex construction of the mitochondrial membranes reflects their functional specialization. Both the OMM and IMM possess transport mechanisms for proteins and metabolites that regulate the structural organization of mitochondria. The OMM is primarily responsible for allocating proteins for specific functions of mitochondria (i.e., protein translocation, mitochondrial quality control, among others) through the regulation of mitophagy and mitochondrial dynamics [8]. In contrast to the OMM, the IMM has a more complex structural organization. It is divided into the inner boundary membrane, a part of the IMM adjacent to the OMM, and the cristae membrane, which forms invaginations projecting into the mitochondrial matrix. Both subdomains communicate through narrow and tubular membrane segments called cristae junctions (CJs), which attach the inner boundary and cristae membranes [9] (Figure 1). Due to heterogeneity in protein composition, membrane structural dynamics, and phospholipid biogenesis, these two membrane domains are morphologically and functionally distinct [9]. The inner boundary membrane, which runs parallel to the OMM, is considered a secondary envelope structure containing protein import machinery close to those present in the OMM, thereby facilitating protein transport into the matrix. The cristae membrane possesses a distinctive fold-like structure that significantly expands its surface area. This topographical feature enables the efficient organization of numerous protein complexes crucial for energy generation through the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS) [10].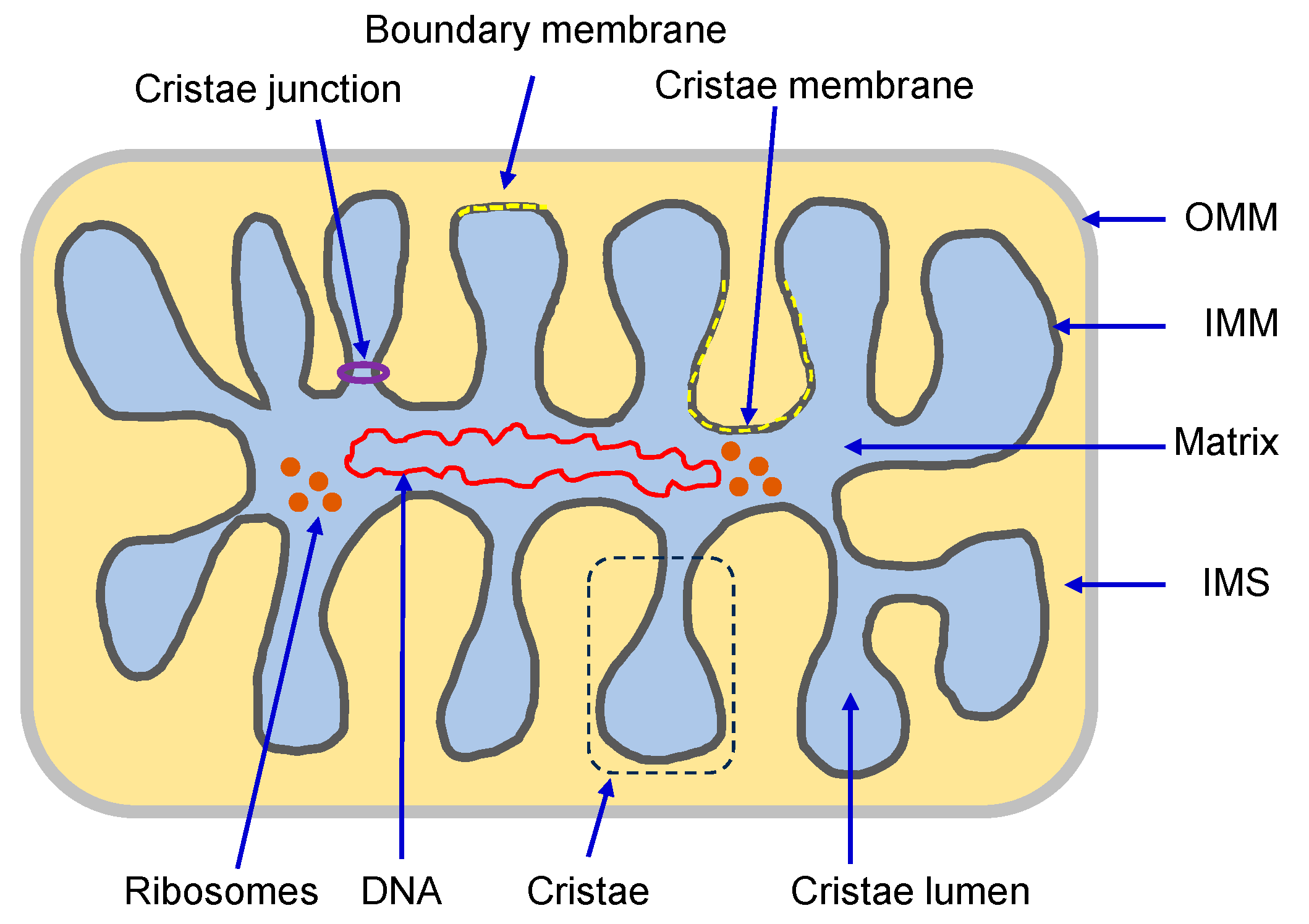
3. Ionic Regulators of Mitochondrial Matrix Volume
Under physiological conditions, ion transport (i.e., Na+, K+, Mg2+, H+, and Ca2+) through the IMM regulates mitochondrial matrix volume [44][45][46]. Slight changes in matrix volume promote mitochondrial activity and stimulate metabolism. Indeed, increases in matrix volume have been shown to stimulate fatty acid oxidation as well as ETC and OXPHOS. Moreover, mitochondrial swelling induces gluconeogenesis by stimulating pyruvate carboxylase [47][48]. Although the exact mechanisms for these effects of mitochondrial swelling are still being elucidated, IMM structural and functional remodeling could be involved in a response against oxidative stress. Hence, numerous oxidative stress-related human disorders, such as cardiovascular and neurological diseases, are influenced by mitochondrial swelling. Among ions, K+ and Ca2+ have been identified as the primary culprits of mitochondria matrix volume changes [45][49]. To keep in line with this, multiple influx and efflux mechanisms are involved in facilitating the transport of K+ and Ca2+ ions across the IMM (Figure 2).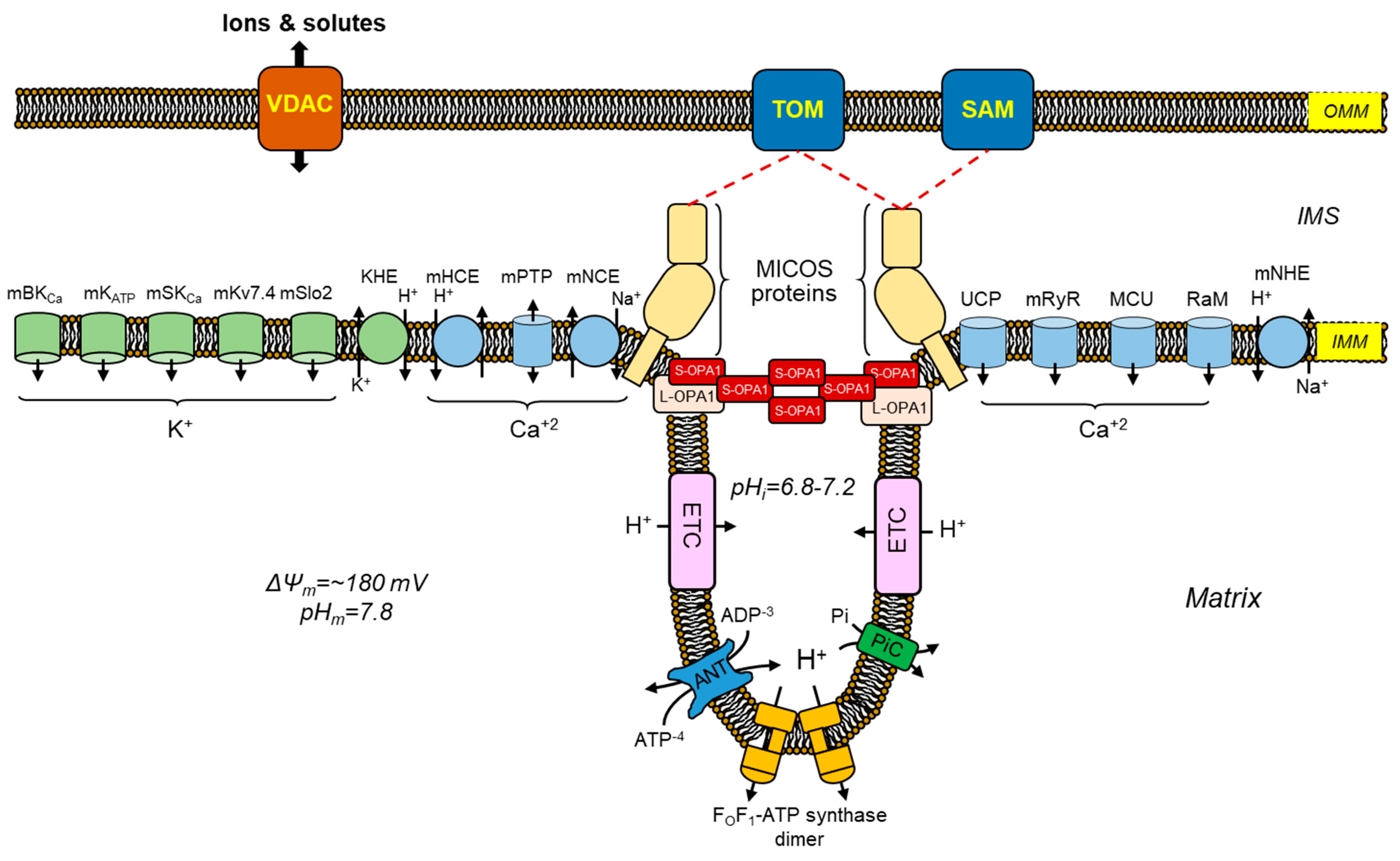
3.1. Transport Mechanisms for Mitochondrial K
+
Mitochondrial physiological swelling is changes in matrix volume that help mediate mitochondrial function and structure. The regulation of mitochondrial matrix volume is a crucial process involving transporting K+ into and out of the matrix. Mitochondrial K+ transport is a complex and dynamic process that affects various cellular activities, such as bioenergetics, Ca2+ regulation, and ROS formation [50]. Therefore, K+ transport could be a target to reduce mitochondrial dysfunction after cardiac IR injury. The identification of molecules that selectively target K+ channels and the lack of information on the significance of each channel to mitochondrial function constitute important obstacles in the development of mitochondrial K+ therapeutics. One K+ efflux and five K+ influx channels have been identified within the IMM of cardiac mitochondria (reviewed in [51]).3.2. Mitochondrial Ca
2+
Transport Mechanisms
3.2.1. Mitochondrial Ca2+ Influx Mechanisms
The cardiac muscle mitochondria harbor three distinct pathways for the influx of Ca2+, namely, the mitochondrial Ca2+ uniporter (MCU), rapid mode of Ca2+ uptake (RaM), and mitochondrial ryanodine receptor (mRyR). Several genetic and pharmacological studies [52][53][54] have established that the MCU is the primary channel for Ca2+ influx in cardiac mitochondria. The molecular constitution of the MCU complex described in 2011 comprises three key components, namely, the MCU, mitochondrial Ca2+ uptake 1–2 (MICU1-2), and essential MCU regulators (EMRE) [55][56][57]. The uptake of Ca2+ through the MCU is significantly dependent on the ΔΨm and is inhibited by Mg2+. The MICU subunits play a role in gatekeeping the MCU complex, regulating Ca2+ entry to prevent mitochondrial Ca2+ overload [58][59]. Studies performed with HEK293T cells recently found a direct interaction between the MCU complex and ETC complex I that helps mediate energy levels when complex I is impaired [60]. Besides regulating bioenergetic homeostasis through complex I, the MCU modulates mitochondrial volume through matrix Ca2+ accumulation. Indeed, impaired matrix Ca2+ accumulation was observed in cardiac mitochondria of MCU knockout mice that could not demonstrate mPTP opening [61]. Furthermore, this study also showed changes in Ca2+-induced mitochondrial swelling in liver, brain, and heart mitochondria that occurred only in the presence of the MCU, further confirming the critical role of MCU in mitochondrial volume regulation and that alternative Ca2+ influx channels only play a small role in regulating this parameter. Pharmacological interventions targeting the MCU have produced favorable cardioprotective outcomes after IR injury. Indeed, the administration of Ru360, a selective inhibitor of the MCU, to rats subjected to an in vivo coronary artery ligation had a protective effect on cardiac and mitochondrial functionality [62]. A similar study conducted on mice involving coronary artery ligation [63] provides additional evidence that the administration of Ru360 reduces the extent of myocardial infarction and preserves the integrity and functionality of mitochondria following IR injury. Currently, the most significant challenges for available MCU-targeting products are their permeability in the cellular membrane and pharmacological specificity [64]. As a result, no clinical studies for these ruthenium compounds have been conducted.3.2.2. Mechanisms for Mitochondrial Ca2+ Efflux
Mitochondria play a critical role in regulating intracellular Ca2+ levels through several transport systems. One of them is the mitochondrial Na+/Ca2+ exchanger (mNCE, also denoted as NCLX or mNCX), an electrogenic transporter that exchanges three cytosolic Na+ for one mitochondrial Ca2+ [65][66]. The mNCE is an important mediator of Ca2+ signaling in cardiac cells, and its dysfunction has been implicated in the pathogenesis of IR injury. The extrusion of mitochondrial Ca2+ during physiological stimulations is limited by mNCE levels, while LETM1 levels are deemed insignificant [67]. For cardiac mitochondria, the mNCE, rather than the mCHE, may be the primary route for mitochondrial Ca2+ efflux [68]. The mNCE protein is a crucial cellular component, and its study offers a valuable understanding of the intricate connections between Ca2+ and redox signaling mechanisms [69]. Upregulation of mNCE provides protection against myocardial IR injury and ischemic heart failure [70]. The same study also demonstrated that the abolition of mNCE leads to left ventricular remodeling, heart failure, and death. These findings suggest that mNCE is essential for normal cardiac function. Although the targeting (activating) of mitochondrial Ca2+ efflux channels for IR injury shows therapeutic potential, there is a lack of pharmacological agents available for this purpose. Therefore, studies indicate that preventing excessive Ca2+ uptake into the mitochondria via MCU inhibition contributes to preserving mitochondrial function and cellular viability. Mitochondrial targeting Ca2+ channel blockers such as ruthenium red products (Ru360 and Ru265) are promising for maintaining cardiac mitochondrial function during Ca2+ overload conditions [71][72]. However, MCU-targeting products are limited due to their membrane permeability properties and pharmacological specificity [64]. In contrast, increasing mitochondrial Ca2+ efflux could be beneficial for preventing mitochondrial injury in pathological conditions [70]. In this line of thought, developing drugs to increase mNCE activity could be promising for treating pathological conditions associated with mitochondrial Ca2+ overload.4. mPTP: A Non-Selective Channel Involved in Mitochondrial Swelling
The mitochondria play a central role in mediating cell death through several pathways, such as apoptosis, ferroptosis, and mPTP-mediated necrosis [73][74]. The occurrence of mPTP-mediated necrosis is attributed to a significant disruption in mitochondrial function in response to energy or oxidative stress. This disruption involves insufficient ATP production, excessive ROS generation, and Ca2+ overload. These conditions attributed to the activation of mPTP lead to cell death. Various factors such as Pi, ADP/ATP, pH fluctuations, Mg2+, and the activation of CypD and Bax/Bak potentially modulate the activity of mPTP [75][76]. The mPTP has been shown to work in both low-conductance (300 pS, reversible) and high-conductance (1.3 nS, irreversible) modes, both of which are reported to contribute to the dissipation of ΔΨm [77]. Also, the opening of mPTP results in substantial swelling, leading to OMM rupture and an increase in ROS production [78]. All factors that contribute to mPTP opening, including mitochondrial Ca2+ overload, ROS accumulation, ATP depletion, high Pi, and ΔΨm loss are present at reperfusion after cardiac ischemia. Therefore, mPTP opening occurs in the reperfused heart and plays a central role in the pathogenesis of cardiac IR (reviewed in [79][80]). Numerous experimental and computational approaches have been employed to study the kinetics of mPTP activation and the shift from a low- to high-conductance state [81][82][83][84]. The utilization of these experimental approaches offers valuable insights into ion homeostasis, facilitating our understanding of the fundamental mechanisms contributing to mitochondrial dysfunction through swelling. Nevertheless, these methodologies have been unsuccessful in identifying the molecular composition of the mPTP. Over the years, there has been a growing interest in investigating mitochondria under both physiological and various pathological conditions (Figure 3A). However, it is noteworthy that the number of studies focusing on the mPTP has shown a declining trend since 2015 (Figure 3B). This decrease in research activity can be attributed to the longstanding challenge of identifying the precise molecular identity and regulatory pathways of the mPTP, which has remained elusive despite extensive research efforts since the 1980s. This lack of clarity has led to a waning interest in this area of study. Recent studies have suggested the possibility of different mPTP forms being formed by ANT or FOF1-ATP synthase, and a potential interaction between these proteins may contribute to their regulatory roles for each other. However, these findings require further investigation and validation. The limited understanding of the molecular structure of the mPTP has hindered the development of new pharmacological compounds targeting this pore. As such, there is a pressing need for additional research to unravel the complexities of the mPTP and identify potential therapeutic targets for drug development. This section discusses the status of current knowledge on the identity of the mPTP and its function in cardiac IR injury.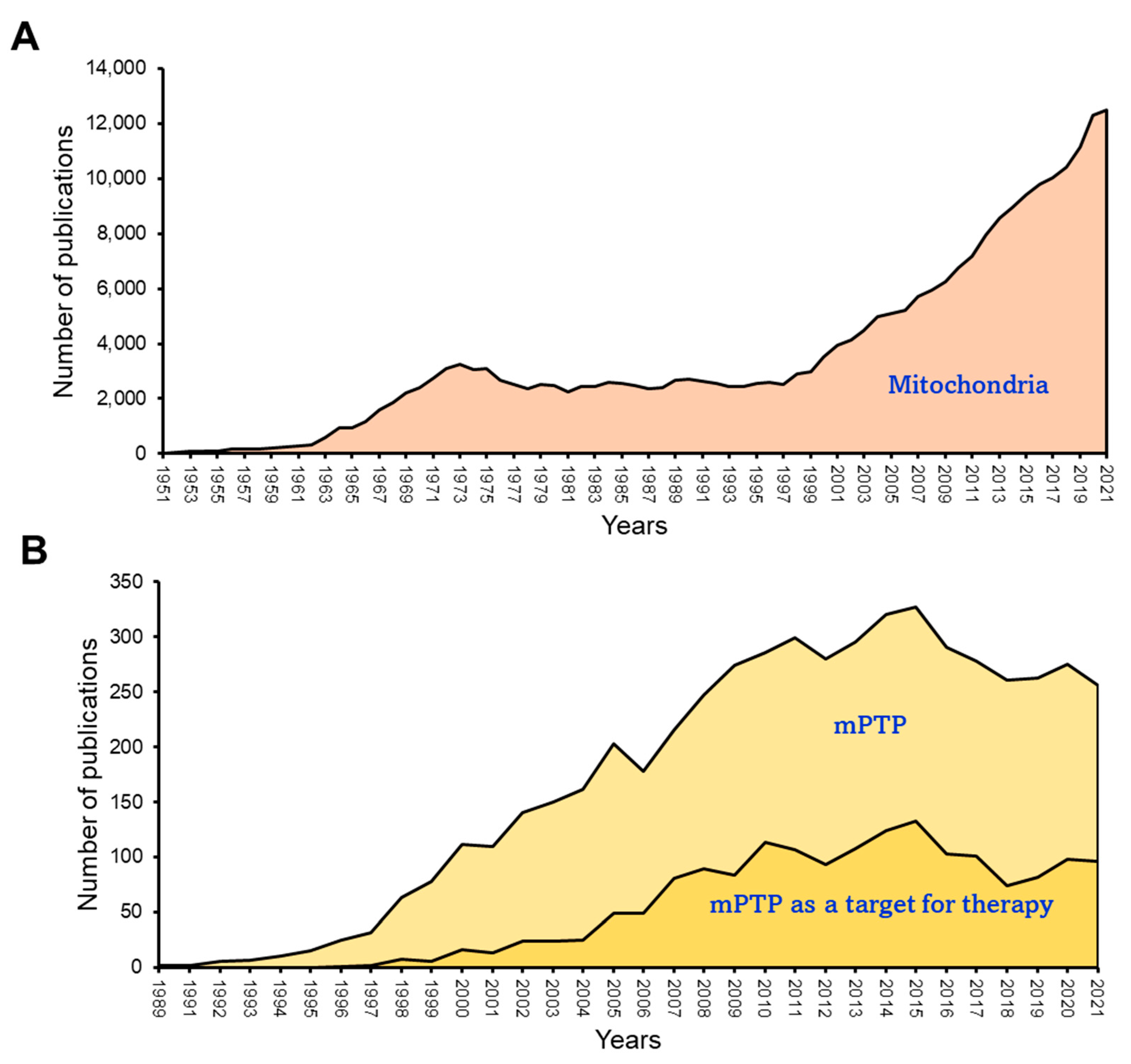
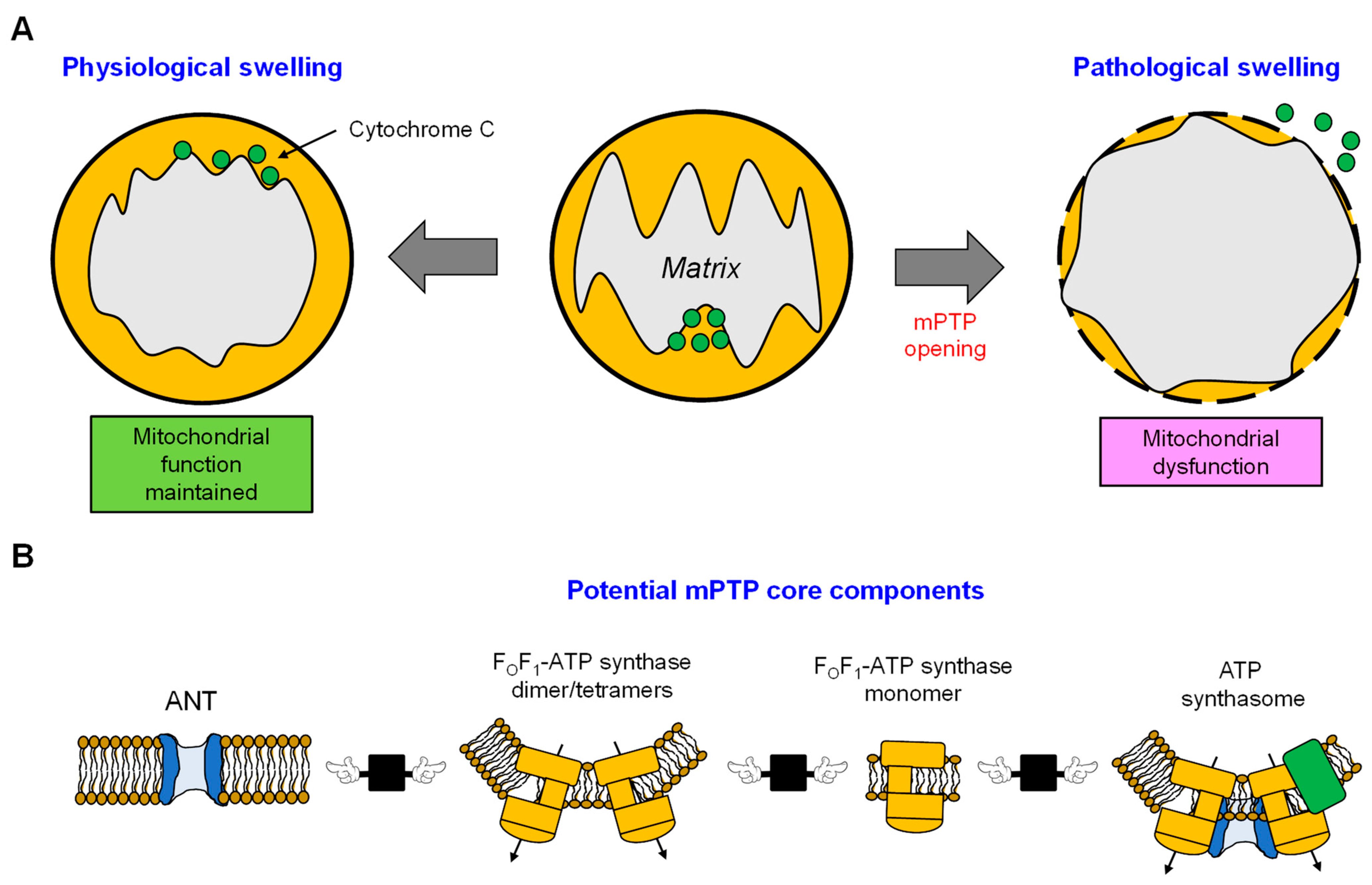
5. The Role of Cytoskeletal Proteins in Mitochondrial Swelling
References
- Ingwall, J.S. Is creatine kinase a target for AMP-activated protein kinase in the heart? J. Mol. Cell Cardiol. 2002, 34, 1111–1120.
- Varikmaa, M.; Guzun, R.; Grichine, A.; Gonzalez-Granillo, M.; Usson, Y.; Boucher, F.; Kaambre, T.; Saks, V. Matters of the heart in bioenergetics: Mitochondrial fusion into continuous reticulum is not needed for maximal respiratory activity. J. Bioenerg. Biomembr. 2013, 45, 319–331.
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454.
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991.
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343.
- Javadov, S.; Chapa-Dubocq, X.; Makarov, V. Different approaches to modeling analysis of mitochondrial swelling. Mitochondrion 2018, 38, 58–70.
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1843, 2233–2239.
- Colina-Tenorio, L.; Horten, P.; Pfanner, N.; Rampelt, H. Shaping the mitochondrial inner membrane in health and disease. J. Intern. Med. 2020, 287, 645–664.
- Vogel, F.; Bornhovd, C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006, 175, 237–247.
- Tarasenko, D.; Meinecke, M. Protein-dependent membrane remodeling in mitochondrial morphology and clathrin-mediated endocytosis. Eur. Biophys. J. 2021, 50, 295–306.
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230.
- Bunik, V.I.; Brand, M.D. Generation of superoxide and hydrogen peroxide by side reactions of mitochondrial 2-oxoacid dehydrogenase complexes in isolation and in cells. Biol. Chem. 2018, 399, 407–420.
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662.
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515.
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707.
- Schenkel, L.C.; Bakovic, M. Formation and regulation of mitochondrial membranes. Int. J. Cell Biol. 2014, 2014, 709828.
- Ball, W.B.; Neff, J.K.; Gohil, V.M. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 2018, 592, 1273–1290.
- Wilson, B.A.; Ramanathan, A.; Lopez, C.F. Cardiolipin-Dependent Properties of Model Mitochondrial Membranes from Molecular Simulations. Biophys. J. 2019, 117, 429–444.
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728.
- de Kroon, A.I.; Dolis, D.; Mayer, A.; Lill, R.; de Kruijff, B. Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim. Biophys. Acta 1997, 1325, 108–116.
- Lu, S.Y.; Graca, T.; Avillan, J.J.; Zhao, Z.; Call, D.R. Microcin PDI Inhibits Antibiotic-Resistant Strains of Escherichia coli and Shigella through a Mechanism of Membrane Disruption and Protection by Homotrimer Self-Immunity. Appl. Environ. Microbiol. 2019, 85, e00371-19.
- Houtkooper, R.H.; Turkenburg, M.; Poll-The, B.T.; Karall, D.; Perez-Cerda, C.; Morrone, A.; Malvagia, S.; Wanders, R.J.; Kulik, W.; Vaz, F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 2009, 1788, 2003–2014.
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 2009, 45, 643–650.
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288.
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia-Reperfusion. Antioxid. Redox Signal. 2017, 27, 57–69.
- Huang, Y.; Powers, C.; Madala, S.K.; Greis, K.D.; Haffey, W.D.; Towbin, J.A.; Purevjav, E.; Javadov, S.; Strauss, A.W.; Khuchua, Z. Cardiac metabolic pathways affected in the mouse model of barth syndrome. PLoS ONE 2015, 10, e0128561.
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schagger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880.
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 2002, 277, 43553–43556.
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 2005, 280, 29403–29408.
- Althoff, T.; Mills, D.J.; Popot, J.L.; Kuhlbrandt, W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664.
- Javadov, S.; Jang, S.; Chapa-Dubocq, X.R.; Khuchua, Z.; Camara, A.K. Mitochondrial respiratory supercomplexes in mammalian cells: Structural versus functional role. J. Mol. Med. 2021, 99, 57–73.
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. Elife 2020, 9, e50973.
- Hackenbrock, C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J. Cell Biol. 1966, 30, 269–297.
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273.
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vazquez, J.; Enriquez, J.A.; Gerle, C.; et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018, 9, 3399.
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67.
- Frezza, C.; Cipolat, S.; de Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189.
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171.
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598.
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646.
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691.
- Gomez-Valades, A.G.; Pozo, M.; Varela, L.; Boudjadja, M.B.; Ramirez, S.; Chivite, I.; Eyre, E.; Haddad-Tovolli, R.; Obri, A.; Mila-Guasch, M.; et al. Mitochondrial cristae-remodeling protein OPA1 in POMC neurons couples Ca(2+) homeostasis with adipose tissue lipolysis. Cell Metab. 2021, 33, 1820–1835.e1829.
- Hu, C.; Shu, L.; Huang, X.; Yu, J.; Li, L.; Gong, L.; Yang, M.; Wu, Z.; Gao, Z.; Zhao, Y.; et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020, 11, 940.
- Halestrap, A.P. Regulation of mitochondrial metabolism through changes in matrix volume. Biochem. Soc. Trans. 1994, 22, 522–529.
- Halestrap, A.P.; Quinlan, P.T.; Whipps, D.E.; Armston, A.E. Regulation of the mitochondrial matrix volume in vivo and in vitro. The role of calcium. Biochem. J. 1986, 236, 779–787.
- Douglas, M.G.; Cockrell, R.S. Mitochondrial cation-hydrogen ion exchange. Sodium selective transport by mitochondria and submitochondrial particles. J. Biol. Chem. 1974, 249, 5464–5471.
- Halestrap, A.P.; Dunlop, J.L. Intramitochondrial regulation of fatty acid beta-oxidation occurs between flavoprotein and ubiquinone. A role for changes in the matrix volume. Biochem. J. 1986, 239, 559–565.
- Owen, M.R.; Halestrap, A.P. The mechanisms by which mild respiratory chain inhibitors inhibit hepatic gluconeogenesis. Biochim. Biophys. Acta 1993, 1142, 11–22.
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163.
- Checchetto, V.; Azzolini, M.; Peruzzo, R.; Capitanio, P.; Leanza, L. Mitochondrial potassium channels in cell death. Biochem. Biophys. Res. Commun. 2018, 500, 51–58.
- Kulawiak, B.; Bednarczyk, P.; Szewczyk, A. Multidimensional Regulation of Cardiac Mitochondrial Potassium Channels. Cells 2021, 10, 1554.
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991.
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486.
- Holmstrom, K.M.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.; Noguchi, A.; et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell. Cardiol. 2015, 85, 178–182.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340.
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature 2020, 582, 129–133.
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 2010, 467, 291–296.
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Wright, L.; Pallafacchina, G.; Terrin, A.; Mammucari, C.; et al. The mitochondrial calcium uniporter (MCU): Molecular identity and physiological roles. J. Biol. Chem. 2013, 288, 10750–10758.
- Balderas, E.; Eberhardt, D.R.; Lee, S.; Pleinis, J.M.; Sommakia, S.; Balynas, A.M.; Yin, X.; Parker, M.C.; Maguire, C.T.; Cho, S.; et al. Mitochondrial calcium uniporter stabilization preserves energetic homeostasis during Complex I impairment. Nat. Commun. 2022, 13, 2769.
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472.
- Garcia-Rivas Gde, J.; Carvajal, K.; Correa, F.; Zazueta, C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837.
- Guan, L.; Che, Z.; Meng, X.; Yu, Y.; Li, M.; Yu, Z.; Shi, H.; Yang, D.; Yu, M. MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy Inhibition. J. Cell. Mol. Med. 2019, 23, 7830–7843.
- Marta, K.; Hasan, P.; Rodriguez-Prados, M.; Paillard, M.; Hajnoczky, G. Pharmacological inhibition of the mitochondrial Ca(2+) uniporter: Relevance for pathophysiology and human therapy. J. Mol. Cell. Cardiol. 2021, 151, 135–144.
- Islam, M.M.; Takeuchi, A.; Matsuoka, S. Membrane current evoked by mitochondrial Na(+)-Ca(2+) exchange in mouse heart. J. Physiol. Sci. 2020, 70, 24.
- Dash, R.K.; Beard, D.A. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 2008, 586, 3267–3285.
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441.
- Haumann, J.; Camara, A.K.S.; Gadicherla, A.K.; Navarro, C.D.; Boelens, A.D.; Blomeyer, C.A.; Dash, R.K.; Boswell, M.R.; Kwok, W.M.; Stowe, D.F. Slow Ca(2+) Efflux by Ca(2+)/H(+) Exchange in Cardiac Mitochondria Is Modulated by Ca(2+) Re-uptake via MCU, Extra-Mitochondrial pH, and H(+) Pumping by F(O)F(1)-ATPase. Front. Physiol. 2018, 9, 1914.
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385.
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 2017, 545, 93–97.
- Ying, W.L.; Emerson, J.; Clarke, M.J.; Sanadi, D.R. Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry 1991, 30, 4949–4952.
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Oh, S.J.; Ikeda, M.; Ide, T.; Hur, K.Y.; Lee, M.S. Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 2022, 8, 414.
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756.
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2013, 2, e00772.
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176.
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831.
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385.
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharmacol. Exp. Ther. 2009, 330, 670–678.
- Baranov, S.V.; Stavrovskaya, I.G.; Brown, A.M.; Tyryshkin, A.M.; Kristal, B.S. Kinetic model for Ca2+-induced permeability transition in energized liver mitochondria discriminates between inhibitor mechanisms. J. Biol. Chem. 2008, 283, 665–676.
- Makarov, V.I.; Khmelinskii, I.; Khuchua, Z.; Javadov, S. In silico simulation of reversible and irreversible swelling of mitochondria: The role of membrane rigidity. Mitochondrion 2020, 50, 71–81.
- Pokhilko, A.V.; Ataullakhanov, F.I.; Holmuhamedov, E.L. Mathematical model of mitochondrial ionic homeostasis: Three modes of Ca2+ transport. J. Theor. Biol. 2006, 243, 152–169.
- Chapa-Dubocq, X.; Makarov, V.; Javadov, S. Simple kinetic model of mitochondrial swelling in cardiac cells. J. Cell. Physiol. 2018, 233, 5310–5321.
- Javadov, S.; Jang, S.; Parodi-Rullan, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia-reperfusion: Whether cyclophilin D is a viable target for cardioprotection? Cell. Mol. Life Sci. 2017, 74, 2795–2813.
- Amanakis, G.; Murphy, E. Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol. 2020, 11, 595.
- Haleckova, A.; Benek, O.; Zemanova, L.; Dolezal, R.; Musilek, K. Small-molecule inhibitors of cyclophilin D as potential therapeutics in mitochondria-related diseases. Med. Res. Rev. 2022, 42, 1822–1855.
- Hausenloy, D.J.; Boston-Griffiths, E.A.; Yellon, D.M. Cyclosporin A and cardioprotection: From investigative tool to therapeutic agent. Br. J. Pharmacol. 2012, 165, 1235–1245.
- Javadov, S.A.; Clarke, S.; Das, M.; Griffiths, E.J.; Lim, K.H.; Halestrap, A.P. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J. Physiol. 2003, 549, 513–524.
- Waldmeier, P.C.; Zimmermann, K.; Qian, T.; Tintelnot-Blomley, M.; Lemasters, J.J. Cyclophilin D as a drug target. Curr. Med. Chem. 2003, 10, 1485–1506.
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799.
- Argaud, L.; Gateau-Roesch, O.; Muntean, D.; Chalabreysse, L.; Loufouat, J.; Robert, D.; Ovize, M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 367–374.
- Fang, J.; Wu, L.; Chen, L. Postconditioning attenuates cardiocyte ultrastructure injury and apoptosis by blocking mitochondrial permeability transition in rats. Acta Cardiol. 2008, 63, 377–387.
- Argaud, L.; Gateau-Roesch, O.; Chalabreysse, L.; Gomez, L.; Loufouat, J.; Thivolet-Bejui, F.; Robert, D.; Ovize, M. Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc. Res. 2004, 61, 115–122.
- Lim, W.Y.; Messow, C.M.; Berry, C. Cyclosporin variably and inconsistently reduces infarct size in experimental models of reperfused myocardial infarction: A systematic review and meta-analysis. Br. J. Pharmacol. 2012, 165, 2034–2043.
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031.
- Neginskaya, M.A.; Morris, S.E.; Pavlov, E.V. Both ANT and ATPase are essential for mitochondrial permeability transition but not depolarization. iScience 2022, 25, 105447.
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17.e12.
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010.
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597.
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988.
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089.
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323.
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465.
- Kwong, J.Q.; Davis, J.; Baines, C.P.; Sargent, M.A.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217.
- Gutierrez-Aguilar, M.; Douglas, D.L.; Gibson, A.K.; Domeier, T.L.; Molkentin, J.D.; Baines, C.P. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell. Cardiol. 2014, 72, 316–325.
- He, L.; Lemasters, J.J. Regulated and unregulated mitochondrial permeability transition pores: A new paradigm of pore structure and function? FEBS Lett. 2002, 512, 1–7.
- Morciano, G.; Naumova, N.; Koprowski, P.; Valente, S.; Sardao, V.A.; Potes, Y.; Rimessi, A.; Wieckowski, M.R.; Oliveira, P.J. The mitochondrial permeability transition pore: An evolving concept critical for cell life and death. Biol. Rev. Camb. Philos. Soc. 2021, 96, 2489–2521.
- Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J. Mol. Cell. Cardiol. 2020, 144, A3–A13.
- Carrer, A.; Tommasin, L.; Sileikyte, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabo, I.; Carraro, M.; et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 2021, 12, 4835.
- Chapa-Dubocq, X.R.; Garcia-Baez, J.F.; Bazil, J.N.; Javadov, S. Crosstalk between adenine nucleotide transporter and mitochondrial swelling: Experimental and computational approaches. Cell Biol. Toxicol. 2022, 39, 435–450.
- Brustovetsky, N.; Klingenberg, M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 1996, 35, 8483–8488.
- Ruck, A.; Dolder, M.; Wallimann, T.; Brdiczka, D. Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS Lett. 1998, 426, 97–101.
- Brustovetsky, N.; Tropschug, M.; Heimpel, S.; Heidkamper, D.; Klingenberg, M. A large Ca2+-dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore. Biochemistry 2002, 41, 11804–11811.
- Boldogh, I.R.; Pon, L.A. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 2006, 1763, 450–462.
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684.
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343.
- Kuznetsov, A.V.; Javadov, S.; Grimm, M.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells 2020, 9, 222.
- Chakrabarti, R.; Ji, W.K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018, 217, 251–268.
- De Vos, K.J.; Allan, V.J.; Grierson, A.J.; Sheetz, M.P. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr. Biol. 2005, 15, 678–683.
- Kuznetsov, A.V.; Javadov, S.; Guzun, R.; Grimm, M.; Saks, V. Cytoskeleton and regulation of mitochondrial function: The role of beta-tubulin II. Front. Physiol. 2013, 4, 82.
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751.
- Illescas, M.; Penas, A.; Arenas, J.; Martin, M.A.; Ugalde, C. Regulation of Mitochondrial Function by the Actin Cytoskeleton. Front. Cell Dev. Biol. 2021, 9, 795838.