1. General Features of Neurodegenerative Diseases
Neurodegenerative diseases have become a major challenge for healthcare systems, as they show a strong correlation with age and life expectancy is rising worldwide
[1]. Their multifactorial origin hampers the discovery of new effective treatments, and current available therapies only procure symptomatic relief to some extent, without modifying the clinical course
[2].
Although some hypotheses have been traditionally proposed to explain the etiopathology of neurodegenerative diseases—e.g., cholinergic hypothesis for Alzheimer´s disease or dopaminergic dysfunction for Parkinson´s disease, recent research in this field attributes these pathologies to several alterations, generally connected to aging and genetic features. Thus, several phenomena such as oxidative stress, proteinopathies, neuroinflammation and excitotoxicity may be the real events causing neurodegeneration and cognitive/motor decline
[3][4][3,4].
Oxidative stress seems to have a key role in neurodegeneration, both by causing direct oxidative damage to lipids, proteins and DNA and by providing feedback to other pathological mechanisms. Oxidative damage of proteins causes them to misfold and aggregate into toxic oligomers or deposits that consequently activate an inflammatory response that aggravates the oxidative status. Mitochondrial DNA also suffers modifications by oxygen and nitrogen reactive species, giving rise to dysfunctions in key proteins involved in cellular metabolism. Thus, mitochondrial activity is impaired, resulting in increased ROS production levels and activation of proapoptotic pathways. Lipid peroxidation by reactive species disrupts the structure and permeability of cellular membranes, leading to inflammation, alterations of calcium homeostasis and neuronal death. These mechanisms are only some representative examples of how oxidative stress plays a central role in the pathological network that underlies neurodegeneration
[5][6][7][5,6,7].
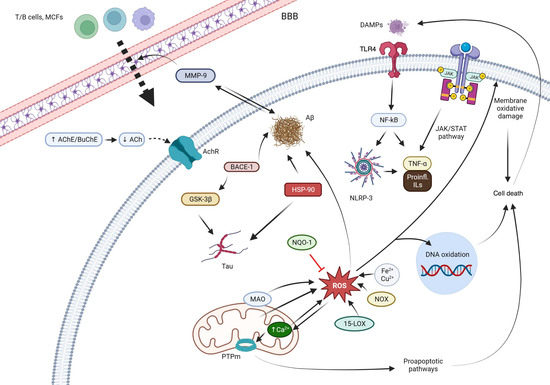
Figure 1 summarizes the pathological mechanisms that are interfered by the quinones.
Figure 1. Main mechanisms of action of the quinones. Abbreviations: T/B cells: the two main types of lymphocytes. BBB: blood brain barrier. AChE: acetylcholinesterase. BChE: butyrylcholinesterase. AChR: acetylcholine receptor. MMP-9: matrix metalloproteinase 9. Aβ: beta-amyloid protein. BACE-1: beta-secretase 1. GSK-3β: glycogen synthase kinase-3 beta. HSP90: Heat-shock protein 90. NQO1: NAD(P)H:quinone oxidoreductase 1. ROS: reactive oxygen species. MAO: monoamino oxidase. PTPm: mitochondrial permeability transition pore. 15-LOX: 15-lipooxygenase. NOX: nitric oxide. DAMPs: damage-associated molecular patterns. TLR4: Toll-like receptor 4. NF-κB: Nuclear factor kappa-light-chain-enhancer of activated B cells. NRLP3: NLR family pyrin domain containing 3. JAK: Janus kinase. STAT: signal transducer and activator of transcription. TNF-α: tumor necrosis factor alpha. IL: interleukins.
Treatments currently employed against neurodegenerative diseases mainly target events such as neurotransmitters deficit, whose alteration is considered nowadays as a consequence of aforementioned phenomena, rather than as the primordial cause of neurodegeneration. Thus, all the efforts are being directed to the discovery of disease-modifying therapies, that may impact on the real causes of these diseases
[8].
Quinones are a family of natural and synthetic compounds characterized by having a fully conjugated cyclic unsaturated dione structure. Quinones have been extensively studied for their biological activities, especially as chemotherapeutic agents
[9][10][11][9,10,11]. Moreover, some endogenous quinones, most notably coenzyme Q
10, show interesting neuroprotective properties, which has prompted the inclusion of quinone structural fragments as a strategy in drug discovery in this area.
2. General Features of Quinone Chemistry Relevant to Neuroprotection
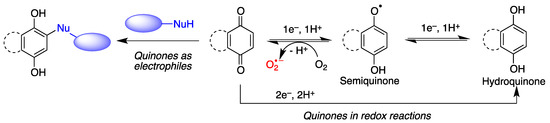
Quinones are often cytoprotective but they can also create a variety of toxic effects, including acute cytotoxicity and immunotoxicity and can therefore be viewed as a double-edged sword in cytoprotection. Although other targets may be involved, these apparently contradictory responses to quinones are generally driven by their chemical properties, summarized in Figure 2.
Figure 2. General features of quinone chemistry of relevance to neuroprotection. Quinones may act as electrophiles, thereby alkylating proteins or nucleic acids. Additionally, they can accept one electron to give semiquinones, which can in turn be reduced to hydroquinones by one-electron transfer and protonation. Semiquinone formation can be reverted by molecular oxygen, leading to the generation of superoxide anion-radicals.
- (a)
-
Quinones are redox active molecules that can form semiquinone radicals by a reversible one-electron transfer process that in the presence of oxygen generates superoxide, a reactive oxygen species in the reverse reaction (Figure 2). Thus, quinones can cause oxidative stress leading to the oxidation of lipids, proteins and DNA.
- (b)
-
Quinones are Michael acceptors and thus can cause cellular damage by alkylating cellular proteins, especially at cysteine residues, or DNA (Figure 2).
- (c)
-
Due to their planar structure and their ability to form hydrogen bonds, many quinones are able to bind to amyloid proteins, preventing their aggregation and allowing the visualization of diffuse and dense-core amyloid plaques
[12][13]. Molecular dynamics simulations have shown that anthraquinone, used as a model simple quinone, interferes with the early phase of aggregation by intercalation into the β-sheet of the hydrophobic H14QKLVFF20 sequence that promotes Aβ self-assembly. The amyloid-quinone complex is formed via polar interactions with the peptide backbone, which destabilize interstrand hydrogen bonds
[13][14].
3. Quinones as Redox Modifiers of Biomolecules
Quinones are pro-oxidant due to their ability to take part in one-electron transfer processes, leading to semiquinones that can then revert to the original quinone by oxidizing a molecule of oxygen to superoxide. This reaction is the basis of the anticancer properties of quinone natural products such as the anthracyclines.
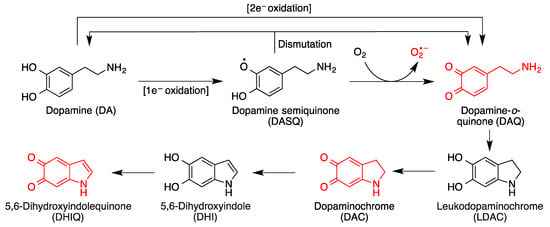
On the other hand, by action of NQO1, an inducible enzyme that catalyzes their two-electron reduction, quinones are transformed into the corresponding antioxidant hydroquinone forms. It is relevant to note that NQO1 levels are increased in patients of diseases that involve high levels of oxidative stress, such as Alzheimer’s disease. In certain situations (e.g., oxidative stress), hydroquinones can be transformed back into quinones, which can lead to cytotoxic responses. One relevant example in the area of neurodegeneration is dopamine, which is a catechol and therefore a hydroquinone (Figure 3). It is the most abundant catecholamine in the brain, and serves as the biosynthetic precursor to other neurotransmitters.
Figure 3. Toxic species derived from the oxidative metabolism of dopamine. Formation of dopamine-o-quinone (DAQ) from the one-electron or two-electron oxidation of dopamine and its evolution by intramolecular Michael additions and additional oxidation reactions.
In situations of oxidative stress such as those prevalent in most neurodegenerative diseases, cytosolic dopamine can generate a variety of neurotoxic species in the brain, including three quinones
[14][15][15,16] (
Figure 3). Dopamine oxidation products act as mitochondrial endotoxins, providing a potential mechanism for preferential neurodegeneration of dopamine-containing neurons in Parkinson’s disease
[16][17], and also have additional toxicities relevant to the onset and progress of Parkinson’s disease, such as impaired protein degradation, mitochondrial dysfunction and α-synuclein aggregation
[17][18].
Other quinones, including environmental toxins such as polychlorinated biphenyl (PCB) quinones have been shown to cause severe cellular oxidative stress
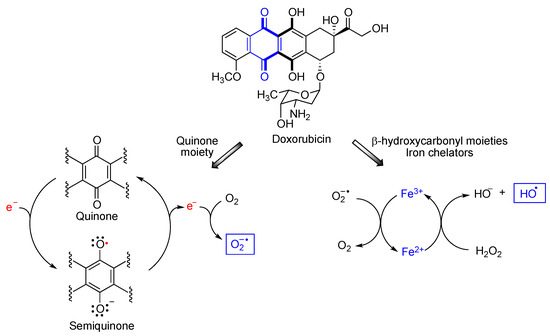
[18][19][19,20]. One particularly interesting case is that of doxorubicin, a member of the anthracycline class of anticancer drugs
[20][21]. Doxorubicin undergoes redox cycling to a semiquinone species that reverts back to the quinone form by one-electron transfer to a molecule of oxygen that becomes superoxide anion, one of the reactive oxygen species (ROS). On the other hand, doxorubicin is also a metal chelator, due to the presence of two γ-hydroxycarbonyl moieties in its structure. Due to ionic interactions with the DNA phosphate groups, the anthracycline-Fe
3+ chelate tightly binds to DNA. When Fe
3+ reacts with the superoxide anion generated by the quinone moiety, it becomes Fe
2+ that forms hydroxyl radicals via its Fenton reaction with hydrogen peroxide (
Figure 4). Doxorubicin causes substantial heart damage due to oxidative stress
[21][22][22,23] and it is also connected to its neurotoxicity, as will be discussed below.
Figure 4.
Mechanisms explaining the generation of reactive oxygen species from doxorubicin.
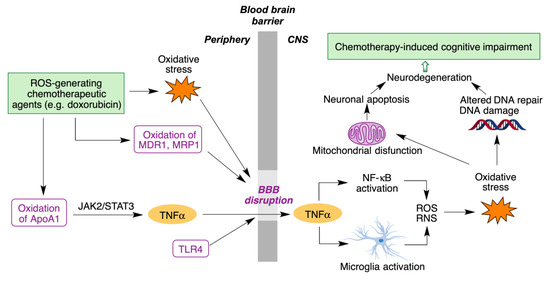
Although doxorubicin cannot cross the blood-brain barrier (BBB), it nevertheless leads to chemotherapy-induced cognitive impairment (CICI), described by patients as “chemobrain” or “chemofog”. CICI is associated with decreased learning abilities, memory and attention capacity that greatly impair the quality of life of many cancer survivors. The complex molecular mechanisms of CICI are initiated by oxidative stress, resulting in protein oxidation and lipid peroxidation, and hence increased levels of the tumor necrosis α factor (TNF-α). On the other hand, activation of the toll-like receptor 4 (TLR4) also contributes to BBB disruption and production of TNF-α and other cytokines. TNF-α is able to cross BBB due to the oxidative stress-mediated disruption of the latter. Once inside the brain, TNF-α triggers local immune response by microglia and NF-κB activation, accompanied by the production of reactive oxygen and nitrogen species (ROS/RN). The subsequent oxidative stress affects DNA repair systems, leading to neurodegeneration. It also affects mitochondria function, with opening of the mitochondria permeability transition pore (mPTP) and release of cytochrome C. This initiates the caspase signaling process, eventually leading to neural apoptosis (
Figure 5)
[23][24].
Figure 5. Main molecular mechanisms involved in chemotherapy-induced cognitive impairment.
4. Quinones as Covalent Modifiers of Biomolecules
Electrophilic compounds, such as quinones, have traditionally had a reputation for toxicity, although covalent drugs are currently undergoing a renaissance and indeed about 30% of the drugs marketed in the last few years belong to this class
[24][25]. As other covalent modulators, quinones can show opposing effects leading to cytoxicity or cytoprotection
[25][26][26,27] depending on the dose, exposure time and intrinsic electrophilicity of the quinone, as shown in
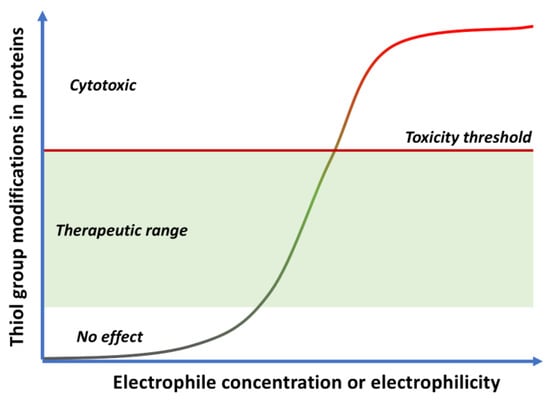
Figure 6.
Figure 6. Cytoxic vs. cytoprotective responses to quinones, depending on their concentration and electrophilicity.
Low doses of moderately electrophilic quinones, which in structural terms are those that are tetrasubstituted or trisubstituted with electron-releasing substituents, are generally cytoprotective. The level of stimulation that they bring about by covalent alteration of biomolecules and by the induction of mitochondrial or cytosolic ROS formation (see
Section 1.2) triggers an adaptive stress response. This response arises mainly from the ability of both electrophiles and oxidants to activate the Keap1/Nrf2 pathway, the key regulator of the phase II antioxidant response, resulting in the induction of the expression of several detoxifying enzymes
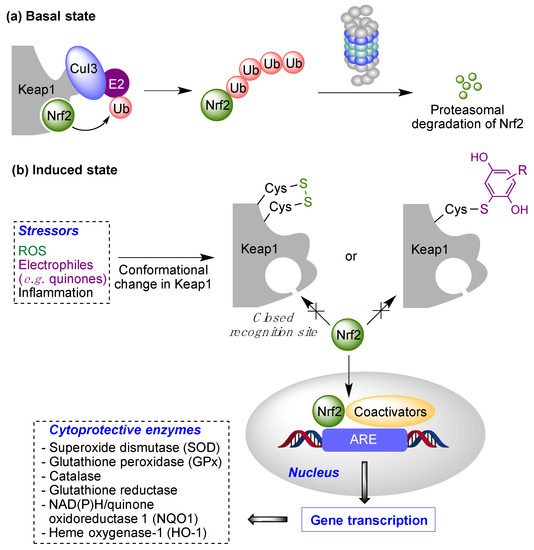
[27][28]. Under physiological, non-stress conditions, Nrf2 is located in the cytosol associated to Keap1 (Kelch-like ECH-associated protein 1), the main negative regulator of Nrf2. Keap1 is able to generate a complex with Cul3, an E3-ubiquitin ligase, inducing Nrf2 ubiquitination and thus its proteasomal degradation
[28][29]. In oxidative stress situations, several cysteine residues (Cys151, Cys273, Cys288 and Cys297) present in the “sensor” region at the C-terminal domain of Keap1 are oxidized to disulfides, which causes a conformational change that releases Nrf2 from Keap1. These cysteine residues can also be activated by reaction with electrophiles, including quinones. After its liberation, Nrf2 translocates into the nucleus, where, after forming heterodimers with several coactivators such as the small musculoaponeurotic fibrosarcoma protein (sMaf), it promotes the transcription of the ARE (antioxidant response element) region in DNA. This region encodes several enzymes able to scavenge ROS and neutralize electrophiles, including superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase, glutathione reductase, NAD(P)H/quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO-1)
[29][30] (
Figure 7). Other protective pathways are known to be activated by quinones, including the heat-shock transcription factor-1 (HSF-1) that induces heat-shock proteins that protect from endoplasmic reticulum (ER)-related stress
[30][31].
Figure 7. The Keap1/Nrf2 cytoprotective pathway. Basal state: Negative regulation of Nrf2 under normal conditions (“basal state”) and its activation under pathological conditions (“induced state” in the presence of reactive oxygen species (ROS) or covalent modifiers.
On the other hand, the administration of more reactive quinones, or high doses of moderately reactive ones, can lead to toxic effects associated to the depletion of glutathione (GSH), a defense mechanism against electrophiles, and the formation of covalent adducts of the quinone to proteins or DNA. In the latter case, long-term exposure may lead to DNA alterations that are not repaired efficiently and can therefore promote mutations and carcinogenesis. Finally, very reactive quinones are likely to react with water before reaching any biomolecule, having therefore little biological effect, or bind only to the oxidative enzymes that generate them (e.g., cytochromes).
One aspect of electrophilic drugs, including quinones, that needs to be addressed at the discovery stage is the possibility that, by reacting indiscriminately against all kinds of proteins, they act as pan-assay interference compounds (PAINS), giving false positives in the drug discovery process
[31][32]. It is nevertheless relevant to note the recent calls for caution against the blind use of PAINS filters
[32][33].