Gypsogenin possesses a versatile and unique aldehyde group that can be utilized to create covalent interactions with undruggable targets. Gypsogenin carboxamides have demonstrated high cytotoxic activity against breast and lung cancer. The bisamides of gypsogenic acid possess prominent activity as well; however, their anti-leukemic activity is yet to be explored.
1. Introduction
Found in higher plants, pentacyclic triterpenes (PTs) are bio-nutrient phytochemicals endowed with a diverse range of bioactivities such as hepatoprotective [1][2][3][17,18,19], anti-inflammatory [4][5][6][20,21,22], anti-hypertensive [3][7][8][9][19,23,24,25], anti-atherosclerotic [7][8][23,24], anti-viral [10][11][12][26,27,28], anti-fibrosis [13][14][15][29,30,31], and anti-ulcer effects [4][16][17][20,32,33]. In particular, PTs have ubiquitous applications in terms of anti-cancer drug discovery [18][19][20][21][22][23][24][25][26][34,35,36,37,38,39,40,41,42].
The literature is loaded with plenty of success stories linking PTs derivatives with a prominent role in the prevention of cancer initiation, promotion, angiogenesis, and progression through disrupting different intermittent mechanisms and pathways. The number of scientific publications and citations linking PTs and cancer has been soaring over the past twenty years, according to the Web of Science database.
PTs comprise four main chemical skeletons, namely oleanane, ursane, lupane, and friedelane. Oleanolic acid, from the oleanane type, is one of the most extensively studied PTs in terms of medicinal chemistry. Oleanolic acid suppresses proliferation of hepatocellular carcinoma
[26][27][42,43], human bladder cancer
[28][44], breast cancer
[29][30][45,46], lung carcer
[31][47], and colon cancer
[32][33][48,49]. To possess such diverse activities, oleanolic acid modulates multiple cell-signaling pathways
[34][50]. Two oleanolic acid derivatives, CDDO and CDDO-Me, have already entered clinical trials for the treatment of solid tumors and lymphoma, allowing oleanolic acid to top the throne of pentacyclic triterpenes in terms of chemotherapy
[35][36][51,52]. Glycyrrhetinic acid is another representative of oleanane-type triterpenoids with ubiquitous anti-cancer activities
[37][38][39][40][41][42][53,54,55,56,57,58]. Ursolic acid
[32][43][44][45][46][48,59,60,61,62], betulinic acid
[47][48][49][50][51][52][63,64,65,66,67,68], and celastrol
[53][54][55][56][69,70,71,72], representing ursane, lupane, and friedelane type triterpenoids, respectively, were reported to possess multifaceted anti-cancer properties.
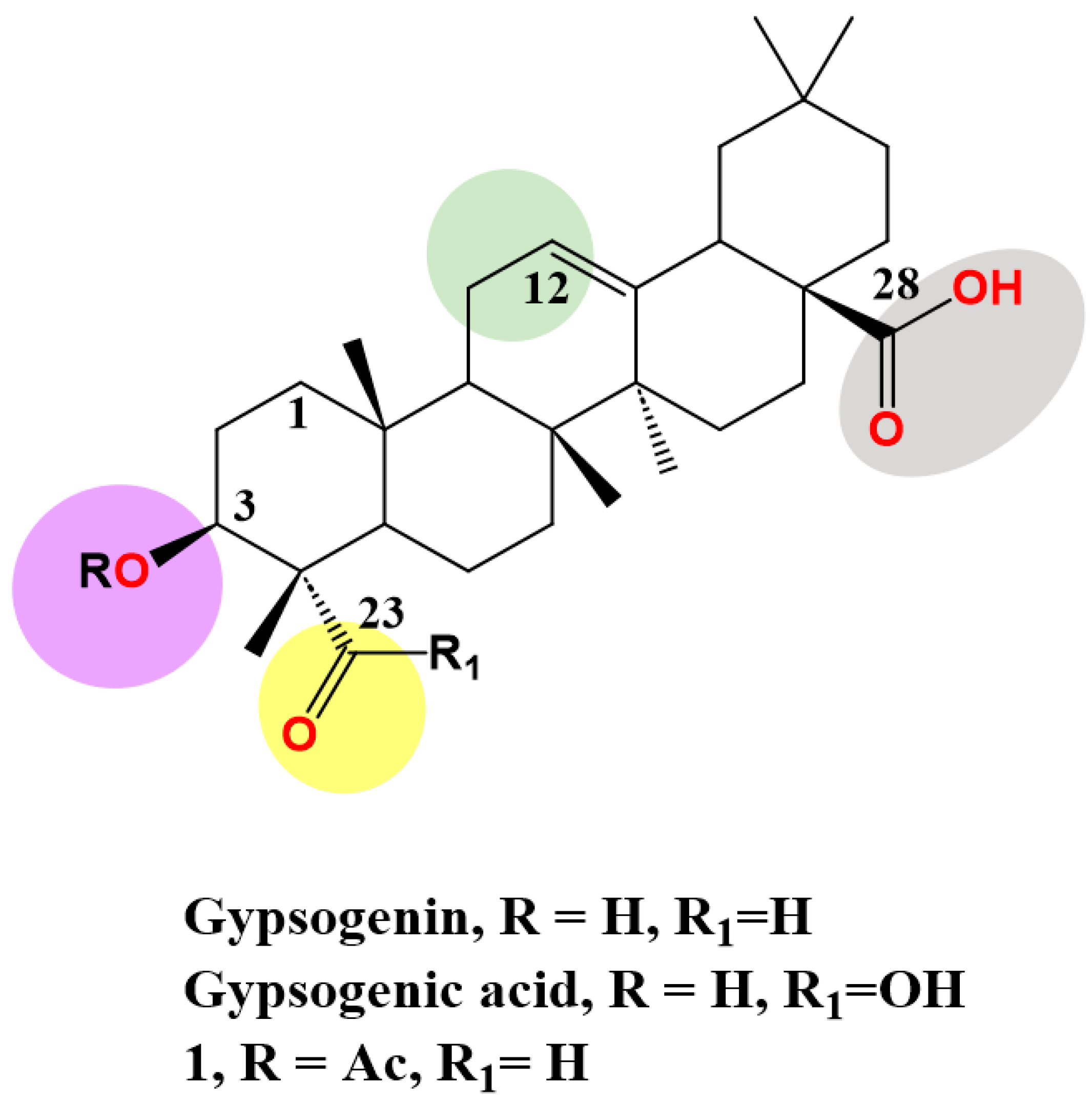
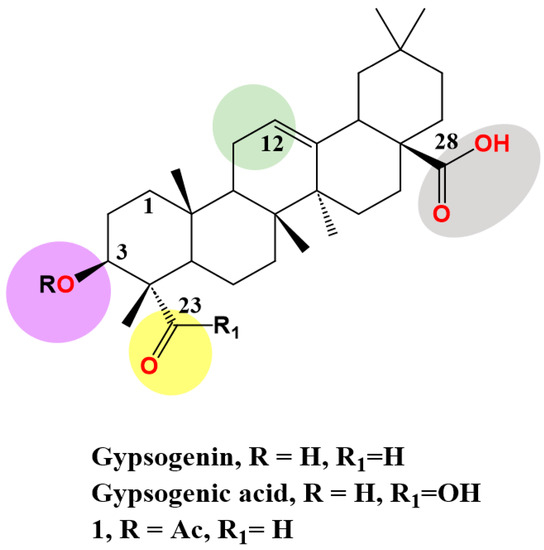
Gypsogenin (3-hydroxy-23-oxoolean-12-en-28-oic acid), a less-explored PT, extracted from
Gypsophila oldhamiana in a saponin form linked with sugar moieties. It is generated as pure sapogenin via acid hydrolysis
[57][73]. It has an oleanane-type skeleton and possesses four active sites, C-3 hydroxyl, ring C double bond, C-23 aldehyde group and C-28 carboxylic acid, which are amenable to a wide range of chemical transformations (
Figure 12). The hydroxyl, alkene, and carboxyl groups exist in most PTs.
Figure 12.
Structure of gypsogenin, gypsogenic acid and 3-acetyl gypsogenin (
1) highlighting the four functional groups.
) highlighting the four functional groups.
2. Anti-Cancer Effect of Gypsogenin, Gypsogenic Acid, and Their Semisynthetic Derivatives
2.1. Anti-Leukemic Activity
Gypsogenic acid did not show observable activity against chronic myeloid leukemia (K562) and acute myeloid leukemia (HL-60), where its IC
50 exceeded 100 µM for both cells
[58][97].
Gypsogenin highly outperforms gypsogenic acid with IC
50 12.7 µM against K562, highlighting the crucial role of the 4-aldehyde group
[59][96]. Simultaneously, Emirdag et al. revealed that gypsogenin has anti-proliferative effect on HL-60 (IC
50 10.4 µM) by inducing apoptosis
[60][61][82,91]. 3-acetyl gypsogenin,
1, has almost the same effect of gypsogenin on HL-60 (IC
50 10.77 µM). Gypsogenin activity is increased by oximation of its aldehyde group (compound
2 IC
50 3.9 µM). Mutually, the 3-acetylated oxime analogue
3 surpassed the activity of
1 (IC
50 5.9 µM)
[61][91]. Gypsogenin benzyl ester
6 has IC
50 8.1 µM; however, its acetylation product
7 has IC
50 6.7 µM
[61] (Figures 2 and 3)[91].
By virtue of its notable apoptotic effect,
6 was further benchmarked for its effect on K562 cell line, where it showed moderate activity (IC
50 9.3 µM)
[62][99]. However, this study represented a turning point for a better understanding of gypsogenin’s molecular target. Compound
6 inhibited ABL1 tyrosine kinase with IC
50 8.71 µM. This is assumed to be the main target for its cytotoxic effect on K562. It is needless to say that the presence of other off targets cannot be excluded. Concomitantly,
6 inhibited other kinases such as C-terminal Src kinase (CSK) and Lyn kinase isoform B; LYN B (IC
50 1.5 µM and 2.9 µM, respectively)
[62][99]. It is clear that oximation of
6 is detrimental for its activity on both K562 and HL-60, as the respective IC
50 value of
4 is 21.3 µM and 10.6 µM
[62][99].
Ciftci et al. moved forward with a structure–activity relationship study of
6 and succeeded in enhancing its activity
[59][96]. As mentioned above, the free aldehyde group is crucial for activity against leukemia. Therefore, Ciftci et al. came up with substituted congeners of 6, keeping a free 4-aldehyde group
[59][96]. Compounds
8 and
9 have IC
50 4.7 and 3.1 µM, respectively, against K562 cells. Additionally, IC
50 of
8 and
9 for ABL1 tyrosine kinase was 7.1 µM and 6.1 µM, respectively.
A recent report by Ulusoy et al. showed that reductive amination of the 4-aldehyde group with different aromatic and alicyclic amines leads to either reduction or complete abrogation of anti-K562 activity
[63][95]. The hit compound in this study,
13, had IC
50 11.3 µM which is even less active than the parent compound, gypsogenin
[63][95]. Furthermore,
13 inhibited ABL1 kinase in a moderate fashion (IC
50 value of 13.0 µM). This is further evidence of the crucial role of the 4-aldehyde group for anti-K562 activity (
Figure 45). In addition,
13 had less effect on MT-2 and Jurkat than
8 and
9. Compound
13 had moderate effect on a panel of kinases at 30 µM of drug concentration, especially for BRK, BTK, LYN B, and SRC. Compound
14 with the more hydrophobic 4-isopropyl substitution exhibited less activity (IC
50 23.8 µM), whereas the presence of a bulky
N-piperazinyl benzyl moiety abolished activity as shown for
12 (IC
50 > 100 µM). The activity was also abolished in the presence of an electron-donating substitution, as was the case for
15 (IC
50 > 100 µM).

Figure 2. Structure of gypsogenin and gypsogenic acid bioactive derivatives through reaction with 3-OH, C-23-CHO or -COOH, and C28-COOH.
Figure 3. Gypsogenin derivatives with modified ring C.
Figure 45.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against K562 and HL-60 cells.
So far, there has been no report linking gypsogenin or gypsogenic acid carboxamides and leukemia. This is the same case for modified ring C derivatives and gypsogenin–chalcone hybrids. In a word, gypsogenin benzyl esters have been the most active derivatives against K562 and HL-60 leukemias until now. The SAR pertaining to activity against K562 and HL-60 is afforded in
Figure 45.
2.2. Anti-Breast Cancer Activity
Gypsogenin has moderate cytotoxic activity for MCF-7 (IC
50 9.0 µM); however, its benzyl ester derivative 6 has IC
50 5.1 µM
[61][91]. Surprisingly, substituted benzyl esters such as 8 and 9 showed less activity than gypsogenin with respective IC
50 51.58 µM and 15.3 µM. Notably, the 3-acetyl analogues 1 and 7 possess less activity (IC
50 20.5 µM and 65.1 µM, respectively). However, oximation of gypsogenin and
6 slightly improves their cytotoxic effect, as shown for 2 and 4. The exact mechanism of action is yet to be elucidated
[61][91]. Notably, compound
1 has low IC
50 value of 5.4 µM against triple-negative breast cancer cell (TNBC) line (MDA-MB-231). In this regard, two gypsogenin–chalcone hybrids demonstrated moderate effect, too, namely,
10 and
11 with respective IC
50 11.0 µM and 7.9 µM
[64][79]. This can be a clue for targeting TNBC, which is an aggressive form of breast cancer that does not respond to hormonal therapy
[65][100].
Wu et al. found that gypsogenic acid has a weak antiproliferative effect on MCF-7 (IC
50 26.8 µM), which also highlights the role of the 4-aldehyde group. The authors highly enhanced gypsogenin and gypsogenic acid activity through mono-and bisamidation
[66][93]. Gypsogenin carboxamide with imidazole, compound
20, has IC
50 3.7 µM, which is similar to the gypsogenic acid mono-amide of only C28 with pyrazole, compound
23, whose IC
50 is 3.8 µM. Gypsogenic acid bisamide of both C23 and C28, compound,
22 demonstrated pronounced activity (IC
50 4.1 µM). The favorable safety profile of those carboxamides is shown by measuring their activity on human umbilical vein endothelial cells (HUVEC cells). It was determined that
22 possesses the highest selectivity index (24.0) among the mentioned active compounds.
Further evidence of the efficiency of gypsogenin amides was disclosed this year by Sun et al.
[67][92]. Two amides,
18 and
19, possess IC
50 5.7 µM and 13.8 µM, respectively, towards MCF-7. They also synthesized compound
16 via reductive amination reaction using methylamine; its IC
50 is 11.3 µM, which is greater than that of gypsogenin (IC
50 9.0 µM). The selectivity index of
16,
18, and
19 exceeds 30 when related to their effect on HUVEC.
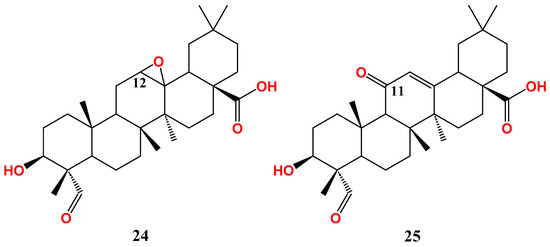
Ring C-oxidized gypsogenin derivatives have recently been developed (
Figure 34)
[67][92]. The epoxide derivative (
24) has IC
50 26.6 µM on MCF-7. In parallel with this, the 11-keto derivative (
25) has similar activity (IC
50 25.3 µM), implying that oxidation of ring C reduces MCF-7 sensitivity. Conclusively, gypsogenin carboxamides are the best cytotoxic entities against MCF-7 compared to other derivatives (
Figure 56).
Figure 56.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against breast cancer cells.
2.3. Anti-Lung Cancer Activity
Gypsogenin can inhibit the growth and metastasis of Lewis lung cancer through inhibition of tumor angiogenesis and induction of apoptosis
[68][101]. Different molecular targets were implicated in this mechanism. Gypsogenin downregulated mutant P53 and vascular endothelial growth factor (VEGF). It reduces the expression of Bcl-2 protein and raises Bax expression, promoting tumor apoptosis. The anti-proliferative effect of gypsogenin, (
1), and 3-acetyl gypsogenic acid against A549 lung cancer cells is moderate (IC
50 19.6, 30.8, and 23.7 µM, respectively)
[57][66][73,93]. Oximation of gypsogenin and
1 maintains the activity without significant change
[57][73]. 2,4-dinitrophenyl)hydrazono derivative of gypsogenin (
5) demonstrated a strong cytotoxic effect on A549 cells (IC
50 3.1 µM)
[57][69][73,80]. In accordance, the amino product (
16) exhibited stronger cytotoxic effect (IC
50 1.5 µM)
[67][92].
The two carboxamides
20 and
23 showed a bit higher activity than compound
5 (IC
50 2.5 and 2.8 µM, respectively)
[66][93]. Both compounds destroyed the cell membrane and increased its permeability, leading to the outflow of intracellular nucleic acid, but they weakly induced apoptosis and arrested A549 cell cycle of
[66][93]. Another anti-lung cancer hit is the gypsogenic acid bisamidation product of (
22), whose IC
50 value is 2.0 µM. However, it is noteworthy that mono-amidation products
20 and
23 surpass its activity but with a lower selectivity index for HUVEC.
Concomitantly, compounds
18 and
19 showed a sub-micromolar effect on A549 (IC
50 0.5 µM and 0.9 µM, respectively) and induced both apoptosis through damaging the cell membrane and arresting the cell cycle. Combining in silico and in vitro tools defined VEGF1 as a gypsogenin target
[67][92]. Remarkably, compound
18 showed a higher binding affinity to VEGF1 than the parent compound, which is in accordance with the cytotoxicity results. Gypsogenin esters showed disappointing results, such as those found for
8, whose IC
50 exceeds 100 µM and
9 which is less active than the parent compound (IC
50 24.5 µM). On the contrary, esterification with chalcone moieties elevated A549 sensitivity; the IC
50 of
10 and
11 is 4.9 µM, and 1.3 µM, respectively
[64][79]. This result denotes the role of chalcone moiety in conferring gypsogenin with high activity.
The epoxide analogue (
24) has almost the same activity as the parent compound (IC
50.18.7 µM), whereas the 11-keto derivative (
25) has slightly better activity (IC
50.13.5 µM)
[67][92]. In conclusion, gypsogenin carboxamides and chalcone hybrids are the most promising anti-proliferative entities against A549 (
Figure 67).
Figure 67.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against lung cancer cells.
2.4. Other Anti-Cancer Activities
A batch of gypsogenin derivatives demonstrated other notable anti-cancer effects. Gypsogenin and its 3-acetyl form (
1) possess remarkable cytotoxic activity against HeLa (cervical cancer)
[64][79]. Compound
2 has notable anti-proliferative activity against SaoS-2 cells (osteosarcoma) and HeLa cells. Its 3-acetylated derivative (
3) also has a similar effect on SaoS-2 but not on HeLa. It is noteworthy that gypsogenin has IC
50 7.8 against SaoS-2 which is better than
1,
2, and
3. On the other hand,
3 is distinguished by its prominent activity against HT-29 cells (colorectal adenocarcinoma)
[61][91].
Another study showed that gypsogenin can suppress gastric cancer cells NCI-N87 proliferation by targeting VEGF and MM-9 and promoting the expression of caspase-3 and Bax proteins
[70][102]. Compounds
5 and
21 were reported mainly for targeting colon cancer cells (LOVO) through strong induction of apoptosis and dose-dependent S-phase arrest in cells. Both compounds exhibited moderate effect on SKOV3 (ovarian cancer) and HepG2 cells (Hepatocellular carcinoma)
[57][73]. The amino compound
16 also exhibited notable activity against LOVO. Compounds
2 and
3 showed no or moderate activity towards LOVO
[67][92].
Three amides were reported by Wu et al.,
20,
22, and
23 with outstanding activities against HepG2, TE-1 (esophageal cancer), and MC3-8 (colon cancer) cells
[66][93]. Gypsogenin–chalcone hybrids
10 and
11 showed outstanding activity against HeLa and pancreatic cancer cells (PANC-1). Gypsogenin 28-COOH ester
9 showed better activity in HeLa cells than
8 [59][96]. Ciftci et al. revealed new derivatives that suppress glioma proliferation through EGFR inhibition. The amino derivative compound
17 has the strongest effect against EGFR and glioma cells U251, T98G, and U87.