Monoclonal antibodies (mAbs), as the name implies, are clonal antibodies that bind to the same antigen. mAbs are broadly used as diagnostic or therapeutic tools for neoplasms, autoimmune diseases, allergic conditions, and infections.
- monoclonal antibodies
- mAbs
- pediatric cancer
1. Introduction
The concept of antibodies dates back to the 18th century when Edward Jenner discovered their role in generating immunity by injecting fluid from a smallpox pustule into recipients [1]. Antibodies (Abs) are glycoproteins belonging to the immunoglobulin superfamily, secreted by plasma cells after B cell lymphocytes encounter foreign antigens. Abs are highly specific and can target various chemical compositions on living/non-living substrates or cell surfaces. Abs are typically produced in labs from animals like rodents, rats, rabbits, goats, horses, camels, or chickens. Using the hybridoma (hybrid between antibody-producing spleen cells and myeloma cells) technique [4], primed clonal B cells can be made immortal thereby continuing to produce mAbs “forever”. By RNA or protein sequencing, the amino acid codes of the binding domains (VH and VL) of any Ab can be defined. With sequences in hand, X-ray crystallography can dissect their 3D structures with precision. In fact, using next-generation sequencing, the entire B cell receptor (BCR) repertoire, i.e., the VH and VL domains of all the B cells can be deduced from a B-cell pool, and the predominant clones (matched VH and VL sequences) predicted using bioinformatics. Today, people can build phage libraries from VH and VL sequences derived from naïve or primed B cells from most species that people can use to fish out mAbs without the need for a living mouse. People even have transgenic rodents carrying human VH and VL sequences that people can immunize to make fully human mAbs. With the current level of sophistication, Abs can be taken apart and built back like legos into forms that fit the purpose of use. Maneuvers such as isotype switch (from IgG1 to IgA), Fc enhancement (to increase FcR affinity), or Fc silencing (to reduce cytokine release syndrome), affinity maturation, multi-specificities, and multi-functionalities are actively being used, and explored. Increasingly, these artificial Ab forms are used to genetically modify the target specificity of both T cells and NK cells in cytotherapy [5][6].
The invention of the hybridoma technique in 1975 [4] marked a milestone in producing mAbs for human use. Since then, over 100 therapeutic mAbs have been authorized by the FDA for various indications in adults[7]. mAbs are clinically valuable in children for both disease diagnosis and treatment. As in vitro diagnostics, they accurately identify normal cells, determine their lineage, and activation status, and detect tumor cells with specific surface antigens like CD3 on T-lymphocytes. Lineage-specific mAbs enable precise diagnosis of immunodeficiencies and hematological malignancies. In treatment, mAbs have shown high efficacy against autoimmune diseases and infectious agents, serving as primary therapy or salvage options for drug-resistant cases[8]. They can be tailored to reduce cytokines, excess Abs, and alloreactive T cells, or enhance immune functions. Classic examples include palivizumab for respiratory syncytial virus [9] and COVID-19 mAbs under emergency use authorization [10][11][12].
2. Monoclonal Antibodies and Pediatric Cancer
Pediatric cancer privileges a survival rate of ≥80% in high-income countries, reaching 95% in acute lymphoblastic leukemia (ALL) or Wilms tumor [11][12][13]. However, the chances of survival are highly variable within tumor entities and among geographic areas in the world. Metastatic or relapsed sarcomas, high-grade brain tumors, and some rare pediatric cancers have a dismal prognosis with no relevant therapeutic advances in decades. Unintended deaths from childhood cancers in low- and middle-income countries, once diagnosed, result from the abandonment of treatment in the setting of complex and intensive treatment regimens, death from toxicity because of insufficient supportive care options, and relapse [12]. The high frequency of long-term sequelae among childhood cancer survivors (nearly 50% with moderate to high multi-organ late effects) [13] has also overshadowed improvement in survival, demanding a reconsideration of treatment intensification typically saddled with toxicities driven to their limits. mAbs are attractive therapeutic alternatives and have already demonstrated efficacy in a variety of childhood cancers [14].
2.1. Monoclonal Antibodies in Pediatric Hematological Malignancies
The first-ever approved mAb for clinical use was rituximab in 1997, a human–mouse chimeric Ab discovered in 1994. Rituximab targets CD20, an antigen expressed on B cell mature hematological malignancies. The phase 3 clinical trial Inter-B-NHL Ritux 2010 (NCT01516580) demonstrated that rituximab added to the standard chemotherapy backbone achieved an event-free survival (EFS) at 3 years of 93.9% compared to 82.3% for the chemotherapy-only group in children with B-cell mature lymphoid malignancies [13]. In 2021, rituximab obtained the approval for pediatric use and when combined with chemotherapy became the first line treatment of high-risk non-Hodgkin lymphoma [14]. Rituximab has a low toxicity profile, mainly with transfusion reactions and hypogammaglobinemia, and is manageable with immunoglobulin replacement. Early studies with gemtuzumab ozogamicin (GO), a humanized anti-CD33 mAb linked to the DNA-binding cytotoxin calicheamicin, showed single-agent activity in refractory pediatric and adult patients with acute myeloid leukemia (AML) (28–30% overall response) [15]. Efficacy and safety in the pediatric population were further supported by data from AAML0531 (NCT00372593), a multicenter randomized study including 1063 patients with newly-diagnosed AML. GO was added to standard chemotherapy in the study arm achieving an estimated percentage of patients free of induction failure, relapse, or death at five years of 48% compared to 40% (95% CI: 36%, 45%) in the chemotherapy arm alone [16]. GO was FDA-approved for the treatment of relapsed or refractory CD33-positive AML in adults and pediatric patients (older than 2 years old) in 2017. Three years later, the FDA extended the indication of GO to newly diagnosed CD33-positive AML to include pediatric patients 1 month and older [17]. In 2011, brentuximab vedotin, an anti-CD30 mAb drug conjugate (ADC) to monomethyl auristatin E, was approved by the FDA in adults for relapsed or refractory Hodgkin lymphoma (HL) and anaplastic large-cell lymphoma (ALCL). In pediatrics, the clinical trial NCT02166463 confirmed a survival advantage among pediatric high-risk HL (EFS at 3 years 92.1% in the brentuximab vedotin group compared to 82.5% for the standard-care group) with low toxicity profile [18], gaining FDA authorization in 2022. In 2014, the FDA granted accelerated approval of blinatumomab for the treatment of Philadelphia chromosome-negative relapsed or refractory precursor B-cell acute lymphoblastic leukemia (R/R B-ALL). Blinatumomab is a bispecific mAb that elicits a cytotoxic T-cell response against CD19-positive cells. On 29 March 2018, the FDA granted accelerated approval of blinatumomab for the treatment of adult and pediatric patients with B-cell precursor ALL in first or second complete remission with minimal residual disease (MRD) greater than or equal to 0.1%. Approval was based on the open-label, multicenter, single-arm BLAST trial (NCT 01207388) [19]. In 2017, the FDA approved inotuzumab ozogamicin for the treatment of adults with relapsed or refractory B-cell precursor ALL. Pediatric authorization is not yet available, but the results are promising in several ongoing and completed clinical trials (NCT02981628, EUDRA-CT 2016-000227-71).2.2. Monoclonal Antibodies Specifically Developed for Pediatric Solid Tumors
Metastatic or relapsed sarcomas, high-grade brain gliomas, and some rare entities (like rhabdoid tumors) are hard to cure, with survival rates below 20% [20]. Treatment of pediatric solid tumors often relies on complex and intense chemotherapy, surgery, and radiotherapy combinations encumbered by significant long-term toxicities. For instance, 93% of high-risk neuroblastoma survivors suffer long-term sequelae, 71% of which are severe, including second malignancies [21]. Today, a variety of mAbs is approved for the treatment of solid tumors in adults, directed against epidermal and vascular growth factors and immune checkpoints [22][23][24][25]. In addition, mAbs in adult oncology are being generated in different formats including antibody-drug conjugates (ADCs) or bispecific T-cell engagers (BiTEs), and targeting a variety of different pro-tumorigenic compounds in the microenvironment or immune checkpoint inhibitors. In contrast, the use of mAbs in pediatric solid tumors in current clinical practice remains anecdotal with one exception, i.e., anti-disialoganglioside mAbs, which are now part of the current standard of care for neuroblastoma.Anti-GD2 Monoclonal Antibodies
Alteration of ganglioside expression in cancer was first reported in 1966 in brain tumors [26] and has since been demonstrated in a large number of human tumors. Disialoganglioside 2 (GD2) is expressed on the outer cell membrane of neural and mesenchymal stem cells during early development. Although GD2 is overexpressed in cancer, its postnatal expression in healthy tissues is restricted to the peripheral neurons, central nervous system, and skin melanocytes [27]. The density of GD2 in neuroblastoma is unusually high, in some estimates could be as high as millions of molecules per cell [28]. High and homogeneous GD2 expression can also be found in subsets of osteosarcoma [29][30][31][32] melanoma cells [33], and some brain tumors [34][35][36]. Other solid tumors such as soft tissue sarcomas, Ewing sarcoma, or desmoplastic small round cell tumor (DRSCT) [37][38] display a lower prevalence and more heterogeneous expression of GD2. Anti-GD2 mAbs bind GD2-expressing tumor cells, engage FcR bearing myeloid effectors to perform Ab-dependent cell-mediated phagocytosis (ADCP), engage FcR-bearing natural killer (NK) cells to perform Ab-dependent cell-mediated cytotoxicity (ADCC), activate complement to perform complement-dependent cytotoxicity (CDC), and, in some instances, cause direct induction of apoptosis [39]. The first-in-man use of anti-GD2 mAb, the murine 3F8 developed in 1985 [40][41] was published in 1987 [33]. After demonstrating its potential in combating marrow disease in patients with primary refractory disease [42], or those in second and first remission [43], the mouse Ab was humanized [39], brought to the clinic in 2011, and became FDA-approved in 2020 [44]. Dinutuximab (ch14.18), an IgG1 human–mouse chimeric switch variant of murine mAb 14G2a was first authorized by the FDA in 2015 in combination with sargramostim a granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2) and 13-cis retinoic acid (RA), for the treatment of pediatric patients with high-risk neuroblastoma with at least partial response after first-line multimodality therapy [45]. The approval was based on results from a phase III, open-label randomized trial conducted by the Children’s Oncology Group (NCT00026312: ANBL0032), where event-free and overall survival were significantly improved among patients with high-risk neuroblastoma that responded to induction therapy, autologous stem cell transplantation, and focal radiotherapy [46]. Dinutuximab beta is a mouse-human chimeric IgG1 mAb produced in a mammalian cell line (CHO) by recombinant DNA technology (ch14.18/CHO). On March 2017, the Committee for Medicinal Products (CHMP) for Human Use adopted a positive opinion, recommending the granting of a marketing authorization under exceptional circumstances for the medicinal product (designated as an orphan medicine in 2012) dinutuximab beta, intended for the treatment of high-risk neuroblastoma in children and adults. The committee also concluded that the active substance contained in dinutuximab beta could not be considered a new active substance. The European Medicines Agency (EMA) approved it for pediatric use in 2017 for the post-consolidation treatment of patients with high-risk neuroblastoma in combination with isotretinoin and IL-2. A randomized phase III study conducted by SIOPEN demonstrated no benefit from the addition of IL-2, which has since been omitted in standard clinical practice [47]. During that decade, naxitamab, the humanized version of m3F8 (hu3F8), received FDA breakthrough designation in 2018 and final approval in 2020 in combination with GM-CSF for pediatric and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow if there is a partial response, minor response, or stable disease to standard induction therapy [44]. Naxitamab has a 10-fold higher affinity than dinutuximab and is humanized rather than chimeric. Even though it is humanized, its immunogenicity after first-time exposure is at least 10%, with a lifetime immunogenicity estimate of 20% after repeated mAb challenges [39]. The FDA approval of naxitamab was based on the results of the pivotal phase II trial (study 201, NCT03363373) in patients with high-risk neuroblastoma in bone and/or bone marrow refractory to initial standard of care or showing insufficient response to therapy for progressive/relapsed disease. The overall response rate (ORR) was 50% (26/52; 95% CI 36–64%) and complete remission (CR) was 38.5% (95% CI 25–53%) [48]. Given GD2 expression in other solid pediatric tumors [37][38][49][50] there may be potential for these Abs in refractory tumors such as osteosarcoma. However, a phase II study carried out by the Children’s Oncology Group (AOST1421) failed to show benefit among patients with recurrent osteosarcoma in complete surgical remission when treated with dinutuximab plus cytokine therapy when compared to historical controls [51]. New clinical trials with naxitamab and dinutuximab beta in osteosarcoma are underway (phase II NCT02502786 and NCT05558280, respectively). Main toxicities from anti-GD2 mAbs are related to GD2 expression by peripheral sensory nerve fibers causing pain in nearly all patients, allergic reactions, myelitis [52], and posterior fossa reversible encephalopathy syndrome (PRES) [53]. No long-term permanent toxicities have been described to date [54].2.3. Monoclonal Antibodies Repurposed for Pediatric Solid Tumors
The first mAb approved for solid tumors was trastuzumab (anti-ERBB2) in 1998 for HER2-positive breast cancer. In children, up to 50% of osteosarcomas express HER2 [55]; however, trastuzumab did not significantly improve survival when combined with chemotherapy in metastatic osteosarcoma [56]. Trastuzumab deruxtecan, an ADC, is being evaluated in HER2-positive osteosarcoma in the PEPN1924 study (NCT04616560). Immune checkpoints (such as PD1 or CTLA4) inhibitors (ICI) have, in a relatively short time, changed the outlook and treatment paradigms for a broad spectrum of adult cancers, complementing or even replacing standard chemotherapy in selected diagnoses as first-line treatment, producing durable remissions not imaginable in the past [24][25]. However, they have limited efficacy in pediatric tumors, except for those with mismatch repair deficiencies [57]. Clinical trials with ICIs have shown limited objective responses in pediatric patients including nivolumab in the ADVL1412 [58] and KEYNOTE-051 [59] for relapsed/refractory solid tumors and pembrolizumab in SARC028 for patients with bone sarcomas [60]. The only promising result so far was seen for atezolizumab, an anti-PD-L1 mAb, among patients with alveolar soft part sarcoma with an ORR of 37% in a phase II study [61]. B7-H3, an immune checkpoint molecule, is overexpressed in multiple cancers including neuroblastoma, sarcomas, and brain tumors [62]. The mAbs 131I-8H9 and 124I-8H9 [63][64], developed against B7-H3, have shown potential in both imaging and treatment of leptomeningeal neuroblastoma [65][66] and diffuse midline gliomas [67]. Different delivery methods (i.e., intraperitoneal, intraOmmaya, or intrapontine) have been employed to avoid hepatic sequestration of the Ab. For a nearly 95% lethal CNS metastasis, 124I-8H9 intraOmmaya treatment yielded a 2-year OS of 57%, EFS > 40% compared to the median 5.5 months survival reported in the literature [68]. However, in a rare disease where randomized arms are not feasible, the lack of comparable historical controls together with safety concerns have prevented its FDA approval for this indication. When combined with abdominopelvic radiotherapy, intraperitoneal radiolabeled 8H9 also increased median overall survival (54 months vs. 34 months) compared to only radiated patients with DSRCT and peritoneal rhabdomyosarcoma [69]. A number of anti-B7-H3 approaches have been undertaken by other groups including naked Fc-enhanced IgG (MGA27) and ADCs [70]. Bevacizumab, an anti-vascular endothelial growth factor (anti-VEGF) mAb, has been studied in various pediatric malignancies with mixed results. It did not show significant benefits in rhabdomyosarcoma [71], osteosarcoma [72], or high-grade glioma [73][74]. Objective responses were observed in low-grade glioma [74] and relapsed/refractory high-risk neuroblastoma [75]. Cetuximab, an anti-EGFR (epidermal growth factor receptor) mAb has neither achieved meaningful responses in pediatric solid tumors [76][77][78][79]. Targeting the insulin-like growth factor-1 receptor (IGF-1R) pathway has shown variable efficacy in children. Ganitumab, an anti-IGF-1R mAb, increased toxicity without improving survival in Ewing sarcoma [80]. Racotumomab, a murine gamma-type anti-idiotype mAb against Neu-glycolyl GM3 ganglioside (NeuGcGM3), overexpressed in some solid pediatric tumors [81], has shown a favorable toxicity profile and immune responses [82]. Its activity in high-risk neuroblastoma is still being evaluated in phase II clinical trial NCT02998983.3. Monoclonal Antibodies for Childhood Cancer: Current Limitations and Future Strategies
The application of mAb therapy in childhood cancer is restricted due to several biological barriers. This section focuses on anti-GD2 mAbs, which have the most clinical experience in pediatric solid tumors.
3.1. Biological Barriers
3.1.1. Limited Clinically Relevant Targets
Pediatric tumors differ significantly from adult cancers as they arise from embryonal cells with distinct genetic and epigenetic aberrations. Pediatric tumors have a low mutational burden, resulting in few neo-antigens and druggable targets. This reduces the frequency of anti-tumor T and B cells and hampers the immune response[83][84]. Targeting alternative tumor-associated antigens, such as GD2, B7H3, L1CAM, GPC3, polysialic acid, DLL3, and HER2, may hold promise for B cell-specific therapies such as mAbs[85][86][87][88]. For B cell targets, tissue distribution of the target is key. The density and the heterogeneity of the target will determine which tumor will escape. Insufficient GD2 density, plus both intratumoral and intertumoral heterogeneity, can account for the failure of anti-GD2 mAbs in tumors other than neuroblastoma (see Figure 1) [47][48][58][59][60].
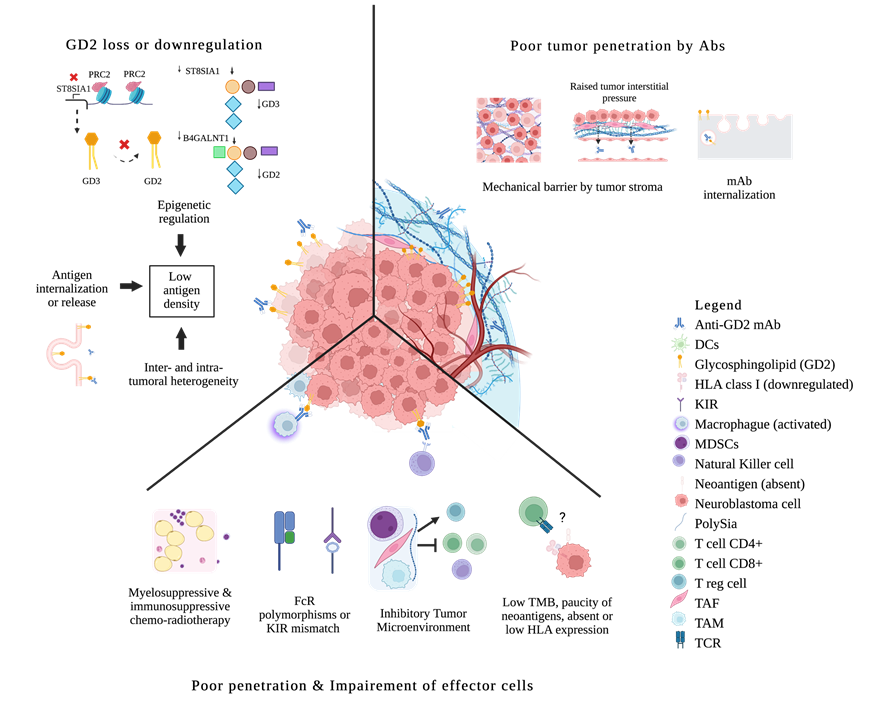
Figure 1. Biologic Resistance to anti-GD2 mAbs in neuroblastoma. Antigen loss or antigen internalization; epigenetic down-regulation of gangliosides synthesis; inter/intratumoral antigen heterogeneity. Poor penetration of mAbs because of mechanical barriers; mAbs disposal through internalization. Impaired effector cells because of chemo-radiotherapy; FcR polymorphisms or KIR mismatch; ineffective tumor infiltration by effector cells due to an inhibitory microenvironment (MDSCs, TAM, TAF); direct immunosuppression by tumors and their released products; paucity of mutations and neoantigens, absent or downregulation of HLA expression, low immunogenicity escaping tumor surveillance. DCs, dendritic cells; FcR, Fc receptor; KIR, killer immunoglobulin-like receptor; MDSCs, myeloid-derived suppressor cells; Polysia, polysialic acid; Treg, regulatory T cells; TAF, tumor-associated fibroblast; TAM, tumor-associated macrophage; TCR, T cell receptor; TMB, tumor mutational burden; ? means absent or downregulation of HLA expression, low immunogenicity escaping tumor surveillance.
3.1.2. Antigen Loss or Downregulation
Repeated exposure to sub-optimal doses of antigen-specific targeting can lead to antigen modulation. Antigens can be lost by release, internalization, or trogocytosis [89] [90][91][1](Figure 1). Acquired genetic alterations as seen in adults, are probably rare in pediatric tumors [3]. Strategies to address this issue include dual-targeting therapies and induction of antigen re-expression. [2][92] (Table 1).
Table 1.
Resistant mechanisms to anti-GD2 therapy and potential alternatives.
| Mechanisms of Anti-GD2 Resistance | Strategies to Overcome Them |
|---|---|
| Antigen loss or downregulation by epigenetic modulation | EZH2 inhibition |
| Poor tumor penetration | Increased payload: Antibody-drug-conjugates Radio-immunotherapy conjugates Drug delivery platforms |
| Impaired effector functions | Fc engineering Engaging T-cells by bispecific antibodies Co-administration with certain cytokines or immuno-conjugates Co-administration with granulocyte-macrophage colony-stimulating factor GM-CSF Early administration of antibodies within the cytotoxic therapeutic plan GD2 conjugated vaccines |
3.1.3. Poor Tumor Penetration
Solid tumors pose challenges for mAb therapy penetration due to leaky vessels, limited lymphatics, altered interstitial pressure, and tumor stroma[93] (Figure 1). Other factors influencing Ab distribution and retention in the tumor include Ab size, affinity, and specificity. Engineered Ab fragments could penetrate better, but their small size below the renal threshold forces their rapid clearance into the urine rendering them sub-therapeutic [94][95]. Higher Ab affinity and higher antigen expression could mitigate the poor retention of small Ab fragments while increasing the cytotoxic payload could amplify the therapeutic effect. Payload optimization has been successful in at least three approaches: (a) drug conjugates, (b) radio-immuno-conjugates, and (c) drug delivery platforms.
a) Drug Conjugates
Drug conjugates, specifically antibody-drug conjugates (ADCs), were developed to increase drug selectivity and reduce unintended systemic toxicity. ADCs utilize antibodies (Abs) to deliver toxic agents precisely to tumor sites, aiming to widen the safety margins between efficacy and side effects. The therapeutic index (TI), which compares drug exposure in tumors to that in normal organs, plays a crucial role. While ADCs show promise, challenges remain, including myelotoxicity and on-target off-tumor effects. Linker chemistry improvements help ensure plasma stability, preventing premature release of cytotoxic payloads. However, the development of anti-drug antibodies (ADA) remains a hurdle when modifying human IgGs. Despite passing efficacy assays, ADCs are expected to encounter toxicities limiting dose escalation in patients [96]. Anti-GD2 ADCs have shown potent cytotoxicity in vitro across various tumor cell lines, but their clinical use will depend on managing toxicity in children [97].
b) Radio-Immuno-Conjugates
Radio-immuno-conjugates use radioisotopes for cancer treatment, but their development has been hindered by suboptimal therapeutic indices and supply chain issues. This limits their application in children. Testing anti-GD2 131I-3F8 in children with metastatic neuroblastoma showed responses in soft tissue and bone marrow, but survival did not improve compared to non-radiolabeled 3F8. Compartmental delivery with intra-Ommaya 131I-3F8 aimed to reduce systemic toxicity and achieved modest success in certain pediatric patients with relapsed neuroblastoma or metastatic medulloblastoma, leading to long-term remissions in some cases (NCT00445965)[98][99].
c) Drug delivery platforms
Refinement of drug delivery platforms remains the key challenge if toxic payloads need further dose escalation to achieve cures. Multi-step Targeting (MST) separates antibody delivery from payload administration, reducing unintended toxicity. Pretargeted strategies (PRIT) using bispecific Abs (BsAbs) offer highly specific targeting, achieving tumor-to-blood ratios not previously possible[100]. BsAbs accumulate in the tumor before payload administration, ensuring precise engagement. Self-assembling and disassembling Abs (SADAs) are designed to stay large in the tumor for penetration and small in the bloodstream to minimize immunogenicity. SADAs successfully deliver radioisotopes without organ toxicity in preclinical models[101]. First human trials of SADA in GD2-positive tumors are ongoing (NCT05130255).
3.1.4. Insufficient or Impaired Effector Functions
Induced host immunity is crucial for durable remission in mAb-treated patients. Anti-idiotypic networks and human antimouse antibodies (HAMA) response may play roles in the anti-tumor response [124][125]. Early administration of anti-GD2 mAbs, combined with induction chemotherapy and immunomodulatory agents, has shown improved objective responses in pediatric patients[126] (Table 1).
3.1.5. Lack of Biomarkers to Predict Response and Survival
The limited success of mAbs in clinical trials may be attributed in part to inadequate patient selection criteria, including risk stratification and target antigen expression. Clinical trials often do not require confirmation of the target antigen expression before enrollment, leading to potential inefficacy in cases where the target antigen is absent or low-density. Biomarkers, such as Fc receptor polymorphisms[127], KIR mismatch[128], and minimal residual disease [129], have been associated with clinical outcomes and response to mAb therapy, but need further validation.
Theranostics, which uses the same mAb for in vivo diagnostic imaging and therapy, has emerged as an appealing drug platform in antibody therapy. Pretargeted radio-immuno-diagnosis is the companion diagnostics for PRIT. It utilizes 177Lu for SPECT and 86Y for PET with high precision; at the same time, 177Lu is used for beta therapy, and 225Ac for alpha therapy [130]. Using whole IgG as a carrier, 68Ga and 64Cu have been used to monitor neuroblastoma during treatment with anti-GD2 [131][132]. Liquid biopsies based on the patient's tumor genotype and circulating GD2 levels have shown potential for detecting residual disease, but further clinical validation is needed at predefined times during treatment[133][134].
3.2 Difficult integration into the standards of care
mAbs exhibit limited antitumor activity as monotherapy, but their efficacy improves when combined with cytokines, chemotherapy, radiotherapy, and kinase inhibitors. Anti-GD2 mAbs show clinical benefits primarily in patients with minimal residual disease (MRD) or exclusive bone/bone marrow involvement, with limited response in soft tissue bulky tumors [11][55][56][57]. However, their combination with chemotherapy, as demonstrated in studies like ANBL1221 and HITS, has proven to be safe and effective for refractory/relapsed neuroblastoma patients. The BEACON study also supports the benefit of adding dinutuximab beta to the standard salvage chemotherapy regimen for relapsed neuroblastoma. Currently, various clinical trials, backed by COG and St. Jude, are investigating the potential benefits of introducing anti-GD2 mAbs in induction therapy[126].
Managing the unique toxicity profile of mAbs poses challenges and requires specialized care teams and dedicated facilities. Visceral pain is a characteristic side effect that demands careful administration and pain management with analgesics and sedation. Efforts to reduce pain side effects through antibody engineering, like the K322A mutation, have had limited success[126]. Prolonged infusion times and desensitization strategies are being explored to minimize side effects and enhance patient acceptance [135][136].
3.3 Commercialization, Regulation, and Political Limitations
Pharmaceutical companies' interest in rare diseases, including pediatric cancer, is growing. However, the profit-driven model often leads to exorbitant drug prices, making mAbs unavailable in developing countries where most children with cancer live (Figure 2). To improve access to life-saving treatments, a cost-driven model similar to handling pandemic vaccines could be adopted, prioritizing need over wealth. Governments can play a crucial role by prioritizing pediatric cancer as a national concern and supporting initiatives that incentivize pharmaceutical companies to invest in pediatric research.
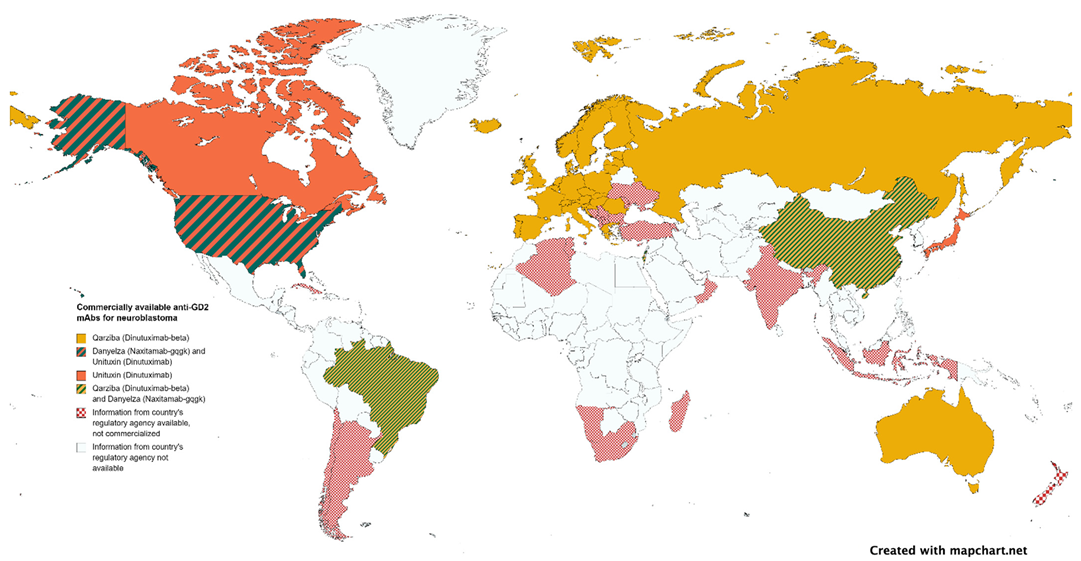
Figure 2.
Authorized anti-GD2 mAbs by national regulatory agencies.
Streamlining regulatory processes and approval procedures is essential to expedite the availability of pediatric drugs, ensuring timely access to life-saving treatments for children [137][138]. Cost-effectiveness considerations must be adapted to suit the pediatric population's unique needs, accounting for long-term consequences and the impact on family members and caregivers.
Innovative approaches such as basket trials based on shared targets and expanded age eligibility are crucial to advance pediatric cancer treatments, given the limited number of available pediatric patients for early-phase trials [139]. Specialized centers of excellence can improve drug delivery efficiency, accessibility, and patient outcomes by unifying care plans and treatment sites, reducing paperwork, and providing training and education for healthcare professionals.
All these barriers are summarized in Figure 3.
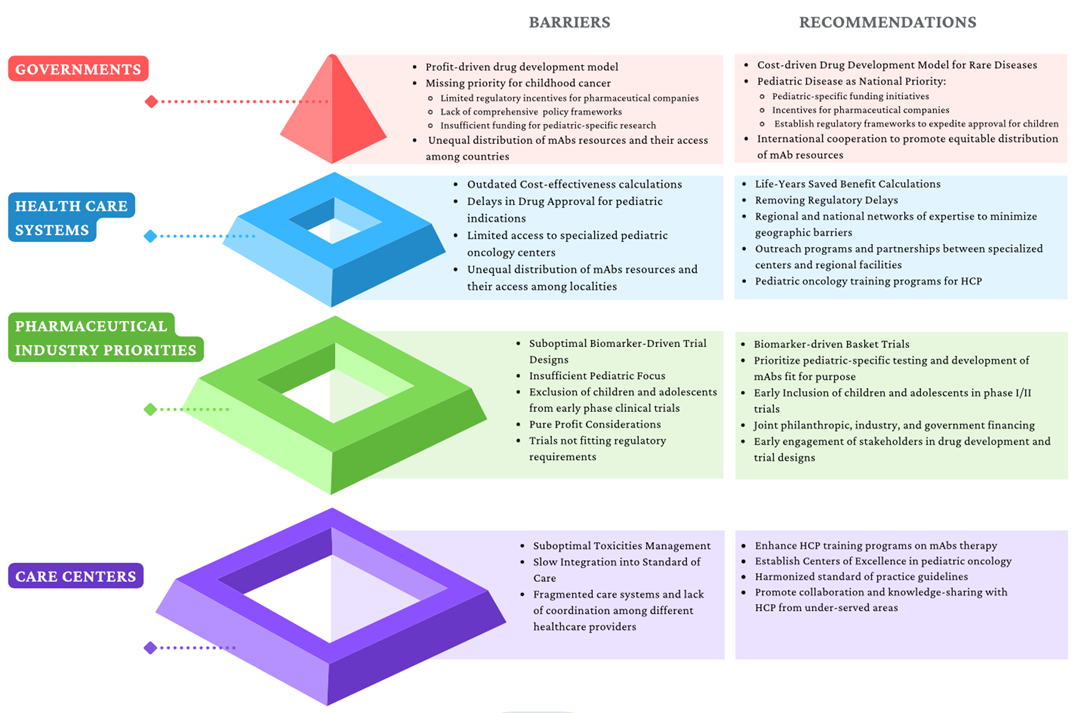
Figure 3.
Identified barriers to mAbs access and proposed recommendations. HCP: health care professionals.
4. Conclusions
Significant progress has been made in mAb-based anticancer therapies in the past decade, and ongoing innovations in protein engineering and immunobiology offer promising avenues for the future. However, challenges persist, including target heterogeneity, tumor microenvironments, and the need for accurate biomarkers. Novel strategies are being explored to overcome these limitations, improving responses and survival in children with cancer.
The projected growth of the mAb market indicates its potential impact on cancer treatment. Academic researchers hold the responsibility to advance new therapeutics, while regulatory agencies should facilitate their translation into clinical practice. Economists and social scientists can promote health policies and international collaboration to maximize the benefits of mAbs as life-changing therapeutics. With concerted efforts, mAbs can bring about transformative change in cancer treatment.
References
- Schoot, R.A.; Otth, M.A.; Frederix, G.W.J.; Leufkens, H.G.M.; Vassal, G. Market Access to New Anticancer Medicines for Children and Adolescents with Cancer in Europe. Eur. J. Cancer 2022, 165, 146–153.
- CADTH Reimbursement Recommendation Dinutuximab (Unituxin) 2. Available online: https://www.cadth.ca/dinutuximab (accessed on 17 April 2023).
- Pritchard-Jones, K.; Pieters, R.; Reaman, G.H.; Hjorth, L.; Downie, P.; Calaminus, G.; Naafs-Wilstra, M.C.; Steliarova-Foucher, E. Sustaining Innovation and Improvement in the Treatment of Childhood Cancer: Lessons from High-Income Countries. Lancet Oncol. 2013, 14, e95–e103.
- Schmitt, J.; Schwenck, J.; Maurer, A.; Przybille, M.; Sonanini, D.; Reischl, G.; Wehrmüller, J.E.; Quintanilla-Martinez, L.; Gillies, S.D.; Krueger, M.A.; et al. Translational ImmunoPET Imaging Using a Radiolabeled GD2-Specific Antibody in Neuroblastoma. Theranostics 2022, 12, 5615–5630.
- Trautwein, N.F.; Reischl, G.; Seitz, C.; Dittmann, H.; Seith, F.; Scheuermann, S.; Feuchtinger, T.; Dombrowski, F.; Handgretinger, R.; Fuchs, J.; et al. First in Human PET/MRI Imaging of in Vivo GD2 Expression in Osteosarcoma. J. Nucl. Med. 2023, 64, 337–338.
- Butch, E.R.; Mead, P.E.; Diaz, V.A.; Tillman, H.; Stewart, E.; Mishra, J.K.; Kim, J.; Bahrami, A.; Dearling, J.L.J.; Packard, A.B.; et al. Positron Emission Tomography Detects In Vivo Expression of Disialoganglioside GD2 in Mouse Models of Primary and Metastatic Osteosarcoma. Cancer Res. 2019, 79, 3112–3124.
- Balis, F.M.; Busch, C.M.; Desai, A.V.; Hibbitts, E.; Naranjo, A.; Bagatell, R.; Irwin, M.; Fox, E. The Ganglioside GD2 as a Circulating Tumor Biomarker for Neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28031.
- Busch, C.M.; Desai, A.V.; Moorthy, G.S.; Fox, E.; Balis, F.M. A Validated HPLC-MS/MS Method for Estimating the Concentration of the Ganglioside, GD2, in Human Plasma or Serum. J. Chromatogr. B 2018, 1102–1103, 60–65.
- Cheung, N.-K.V.; Kushner, B.H.; Cheung, I.Y.; Kramer, K.; Canete, A.; Gerald, W.; Bonilla, M.A.; Finn, R.; Yeh, S.J.; Larson, S.M. Anti-GD 2 Antibody Treatment of Minimal Residual Stage 4 Neuroblastoma Diagnosed at More Than 1 Year of Age. J. Clin. Oncol. 1998, 16, 3053–3060.
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334.
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with Anti-GD2 Antibody Ch14.18/CHO (Dinutuximab Beta) in Patients with High-Risk Neuroblastoma (HR-NBL1/SIOPEN): A Multicentre, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1617–1629.
- Mora, J.; Bear, M.; Chan, G.; Morgenstern, D.A.; Nysom, K.; Tornøe, K.; Sørensen, P.S.; Kushner, B. 891P Naxitamab Treatment for Relapsed or Refractory High-Risk Neuroblastoma: Outcomes from the First Prespecified Analyses of the Pivotal 201 Trial. Ann. Oncol. 2022, 33, S956.
- Mora, J.; Chan, G.C.; Morgenstern, D.A.; Nysom, K.; Bear, M.K.; Tornøe, K.; Kushner, B.H. Outpatient Administration of Naxitamab in Combination with Granulocyte-macrophage Colony-stimulating Factor in Patients with Refractory and/or Relapsed High-risk Neuroblastoma: Management of Adverse Events. Cancer Rep. 2023, 6, e1627.
- Varo, A.; Castañeda, A.; Chamorro, S.; Muñoz, J.P.; Gorostegui, M.; Celma, M.S.; Lopez, S.; Simao, M.; Perez-Jaume, S.; Mora, J. Novel Infusion Strategy Reduces Severe Adverse Events Caused by the Anti-GD2 Monoclonal Antibody Naxitamab. Front. Oncol. 2023, 13, 1164949.
- Oganesyan, V.; Damschroder, M.M.; Leach, W.; Wu, H.; Dall’Acqua, W.F. Structural Characterization of a Mutated, ADCC-Enhanced Human Fc Fragment. Mol. Immunol. 2008, 45, 1872–1882.
- Espinosa-Cotton, M.; Cheung, N.K.V. Bispecific Antibodies for the Treatment of Neuroblastoma. Pharmacol. Ther. 2022, 237, 108241.
- Aldoss, I.; Bargou, R.C.; Nagorsen, D.; Friberg, G.R.; Baeuerle, P.A.; Forman, S.J. Redirecting T Cells to Eradicate B-Cell Acute Lymphoblastic Leukemia: Bispecific T-Cell Engagers and Chimeric Antigen Receptors. Leukemia 2017, 31, 777–787.
- Algeri, M.; Del Bufalo, F.; Galaverna, F.; Locatelli, F. Current and Future Role of Bispecific T-Cell Engagers in Pediatric Acute Lymphoblastic Leukemia. Expert. Rev. Hematol. 2018, 11, 945–956.
- Ribera, J.-M.; Genescà, E.; Ribera, J. Bispecific T-Cell Engaging Antibodies in B-Cell Precursor Acute Lymphoblastic Leukemias: Focus on Blinatumomab. Ther. Adv. Hematol. 2020, 11, 2040620720919632.
- Lopez-Albaitero, A.; Xu, H.; Guo, H.; Wang, L.; Wu, Z.; Tran, H.; Chandarlapaty, S.; Scaltriti, M.; Janjigian, Y.; de Stanchina, E.; et al. Overcoming Resistance to HER2-Targeted Therapy with a Novel HER2/CD3 Bispecific Antibody. Oncoimmunology 2017, 6, e1267891.
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The Absence of Fucose but Not the Presence of Galactose or Bisecting N-Acetylglucosamine of Human IgG1 Complex-Type Oligosaccharides Shows the Critical Role of Enhancing Antibody-Dependent Cellular Cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473.
- Nguyen, R.; Moustaki, A.; Norrie, J.L.; Brown, S.; Akers, W.J.; Shirinifard, A.; Dyer, M.A. Interleukin-15 Enhances Anti-GD2 Antibody-Mediated Cytotoxicity in an Orthotopic PDX Model of Neuroblastoma. Clin. Cancer Res. 2019, 25, 7554–7564.
- Cheung, N.-K.V.; Cañete, A.; Cheung, I.Y.; Ye, J.-N.; Liu, C. Disialoganglioside g d 2 anti-idiotypic monoclonal antibodies. Int. J. Cancer 1993, 54, 499–505.
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.-K. Humanized 3F8 Anti-G2 Monoclonal Antibody Dosing with Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma. JAMA Oncol. 2018, 4, 1729.
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334.
- Ozkaynak, M.F.; Gilman, A.L.; London, W.B.; Naranjo, A.; Diccianni, M.B.; Tenney, S.C.; Smith, M.; Messer, K.S.; Seeger, R.; Reynolds, C.P.; et al. A Comprehensive Safety Trial of Chimeric Antibody 14.18 with GM-CSF, IL-2, and Isotretinoin in High-Risk Neuroblastoma Patients Following Myeloablative Therapy: Children’s Oncology Group Study ANBL0931. Front. Immunol. 2018, 9, 1355.
- Ladenstein, R.L.; Poetschger, U.; Valteau-Couanet, D.; Gray, J.; Luksch, R.; Balwierz, W.; Castel, V.; Ash, S.; Popovic, M.B.; Laureys, G.; et al. Randomization of Dose-Reduced Subcutaneous Interleukin-2 (ScIL2) in Maintenance Immunotherapy (IT) with Anti-GD2 Antibody Dinutuximab Beta (DB) Long-Term Infusion (LTI) in Front–Line High-Risk Neuroblastoma Patients: Early Results from the HR-NBL1/SIOPEN Trial. J. Clin. Oncol. 2019, 37, 10013.
- Park, J.R.; Scott, J.R.; Stewart, C.F.; London, W.B.; Naranjo, A.; Santana, V.M.; Shaw, P.J.; Cohn, S.L.; Matthay, K.K. Pilot Induction Regimen Incorporating Pharmacokinetically Guided Topotecan for Treatment of Newly Diagnosed High-Risk Neuroblastoma: A Children’s Oncology Group Study. J. Clin. Oncol. 2011, 29, 4351–4357.
- Cheung, I.Y.; Cheung, N.K.V.; Modak, S.; Mauguen, A.; Feng, Y.; Basu, E.; Roberts, S.S.; Ragupathi, G.; Kushner, B.H. Survival Impact of Anti-GD2 Antibody Response in a Phase II Ganglioside Vaccine Trial Among Patients With High-Risk Neuroblastoma With Prior Disease Progression. J. Clin. Oncol. 2021, 39, 215–226.
- Cheung, N.-K.V.; Sowers, R.; Vickers, A.J.; Cheung, I.Y.; Kushner, B.H.; Gorlick, R. FCGR2A Polymorphism Is Correlated With Clinical Outcome After Immunotherapy of Neuroblastoma With Anti-GD2 Antibody and Granulocyte Macrophage Colony-Stimulating Factor. J. Clin. Oncol. 2006, 24, 2885–2890.
- Cheung, N.K.; Lazarus, H.; Miraldi, F.D.; Abramowsky, C.R.; Kallick, S.; Saarinen, U.M.; Spitzer, T.; Strandjord, S.E.; Coccia, P.F.; Berger, N.A. Ganglioside GD2 Specific Monoclonal Antibody 3F8: A Phase I Study in Patients with Neuroblastoma and Malignant Melanoma. J. Clin. Oncol. 1987, 5, 1430–1440.
- Wikstrand, C.J.; Fredman, P.; Svennerholm, L.; Humphrey, P.A.; Bigner, S.H.; Bigner, D.D. Monoclonal Antibodies to Malignant Human Gliomas. Mol. Chem. Neuropathol. 1992, 17, 137–146.
- Mennel, H.D.; Bosslet, K.; Geissel, H.; Bauer, B.L. Immunohistochemically Visualized Localisation of Gangliosides Glac2 (GD3) and Gtri2 (GD2) in Cells of Human Intracranial Tumors. Exp. Toxicol. Pathol. 2000, 52, 277–285.
- Shinoura, N.; Dohi, T.; Kondo, T.; Yoshioka, M.; Takakura, K.; Oshima, M. Ganglioside Composition and Its Relation to Clinical Data in Brain Tumors. Neurosurgery 1992, 31, 541–549.
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000.
- Chang, H.R.; Cordon-Cardo, C.; Houghton, A.N.; Cheung, N.-K.V.; Brennan, M.F. Expression of Disialogangliosides GO2 and GD3 on Human Soft Tissue Sarcomas. Cancer 1992, 70, 633–638.
- Cheung, N.-K.K.V.; Guo, H.; Hu, J.; Tassev, D.V.; Cheung, I.Y. Humanizing Murine IgG3 Anti-GD2 Antibody M3F8 Substantially Improves Antibody-Dependent Cell-Mediated Cytotoxicity While Retaining Targeting in Vivo. Oncoimmunology 2012, 1, 477–486.
- Saito, M.; Yu, R.K.; Cheung, N.-K.V. Ganglioside GD2 Specificity of Monoclonal Antibodies to Human Neuroblastoma Cell. Biochem. Biophys. Res. Commun. 1985, 127, 1–7.
- Cheung, N.K.; Saarinen, U.M.; Neely, J.E.; Landmeier, B.; Donovan, D.; Coccia, P.F. Monoclonal Antibodies to a Glycolipid Antigen on Human Neuroblastoma Cells. Cancer Res. 1985, 45, 2642–2649.
- Cheung, N.-K.V.; Kushner, B.H.; Cheung, I.Y.; Kramer, K.; Canete, A.; Gerald, W.; Bonilla, M.A.; Finn, R.; Yeh, S.J.; Larson, S.M. Anti-GD 2 Antibody Treatment of Minimal Residual Stage 4 Neuroblastoma Diagnosed at More Than 1 Year of Age. J. Clin. Oncol. 1998, 16, 3053–3060.
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.-K. Humanized 3F8 Anti-G2 Monoclonal Antibody Dosing with Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma. JAMA Oncol. 2018, 4, 1729.
- Markham, A. Naxitamab: First Approval. Drugs 2021, 81, 291–296.
- Dhillon, S. Dinutuximab: First Global Approval. Drugs 2015, 75, 923–927.
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334.
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with Anti-GD2 Antibody Ch14.18/CHO (Dinutuximab Beta) in Patients with High-Risk Neuroblastoma (HR-NBL1/SIOPEN): A Multicentre, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1617–1629.
- Mora, J.; Bear, M.; Chan, G.; Morgenstern, D.A.; Nysom, K.; Tornøe, K.; Sørensen, P.S.; Kushner, B. 891P Naxitamab Treatment for Relapsed or Refractory High-Risk Neuroblastoma: Outcomes from the First Prespecified Analyses of the Pivotal 201 Trial. Ann. Oncol. 2022, 33, S956.
- Navid, F.; Santana, V.M.; Barfield, R.C. Anti-GD2 Antibody Therapy for GD2-Expressing Tumors. Curr. Cancer Drug. Targets 2010, 10, 200–209.
- Dobrenkov, K.; Ostrovnaya, I.; Gu, J.; Cheung, I.Y.; Cheung, N.K.V. Oncotargets GD2 and GD3 Are Highly Expressed in Sarcomas of Children, Adolescents, and Young Adults. Pediatr. Blood Cancer 2016, 63, 1780–1785.
- Hingorani, P.; Krailo, M.; Buxton, A.; Hutson, P.; Sondel, P.M.; Diccianni, M.; Yu, A.; Morris, C.D.; Womer, R.B.; Crompton, B.; et al. Phase 2 Study of Anti-Disialoganglioside Antibody, Dinutuximab, in Combination with GM-CSF in Patients with Recurrent Osteosarcoma: A Report from the Children’s Oncology Group. Eur. J. Cancer 2022, 172, 264–275.
- Ding, Y.Y.; Panzer, J.; Maris, J.M.; Castañeda, A.; Gomez-Chiari, M.; Mora, J. Transverse Myelitis as an Unexpected Complication Following Treatment with Dinutuximab in Pediatric Patients with High-Risk Neuroblastoma: A Case Series. Pediatr. Blood Cancer 2018, 65, e26732.
- Kushner, B.H.; Modak, S.; Basu, E.M.; Roberts, S.S.; Kramer, K.; Cheung, N.K.V. Posterior Reversible Encephalopathy Syndrome in Neuroblastoma Patients Receiving Anti-GD2 3F8 Monoclonal Antibody. Cancer 2013, 119, 2789–2795.
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-Term Follow-up of a Phase III Study of Ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer Res. 2021, 27, 2179–2189.
- Wang, S.; Zhong, G.; Wang, X.; Yu, F.; Weng, D.; Wang, X.; Lin, J. Prognostic Significance of the Expression of HER Family Members in Primary Osteosarcoma. Oncol. Lett. 2018, 16, 2185–2194.
- Ebb, D.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II Trial of Trastuzumab in Combination with Cytotoxic Chemotherapy for Treatment of Metastatic Osteosarcoma with Human Epidermal Growth Factor Receptor 2 Overexpression: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 2545–2551.
- Lee, V.; Murphy, A.; Le, D.T.; Diaz, L.A. Mismatch Repair Deficiency and Response to Immune Checkpoint Blockade. Oncologist 2016, 21, 1200–1211.
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in Children and Young Adults with Relapsed or Refractory Solid Tumours or Lymphoma (ADVL1412): A Multicentre, Open-Label, Single-Arm, Phase 1–2 Trial. Lancet Oncol. 2020, 21, 541–550.
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.S.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. KEYNOTE-051: An Update on the Phase 2 Results of Pembrolizumab (Pembro) in Pediatric Patients (Pts) with Advanced Melanoma or a PD-L1–Positive Advanced, Relapsed or Refractory Solid Tumor or Lymphoma. J. Clin. Oncol. 2018, 36, 10525.
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in Advanced Soft-Tissue Sarcoma and Bone Sarcoma (SARC028): A Multicentre, Two-Cohort, Single-Arm, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1493–1501.
- Naqash, A.R.; O’Sullivan Coyne, G.H.; Moore, N.; Sharon, E.; Takebe, N.; Fino, K.K.; Ferry-Galow, K.V.; Hu, J.S.; Van Tine, B.A.; Burgess, M.A.; et al. Phase II Study of Atezolizumab in Advanced Alveolar Soft Part Sarcoma (ASPS). J. Clin. Oncol. 2021, 39, 11519.
- Modak, S.; Kramer, K.; Gultekin, S.H.; Guo, H.F.; Cheung, N.K. Monoclonal Antibody 8H9 Targets a Novel Cell Surface Antigen Expressed by a Wide Spectrum of Human Solid Tumors. Cancer Res. 2001, 61, 4048–4054.
- Loo, D.; Alderson, R.F.; Chen, F.Z.; Huang, L.; Zhang, W.; Gorlatov, S.; Burke, S.; Ciccarone, V.; Li, H.; Yang, Y.; et al. Development of an Fc-Enhanced Anti–B7-H3 Monoclonal Antibody with Potent Antitumor Activity. Clin. Cancer Res. 2012, 18, 3834–3845.
- Kramer, K.; Cheung, N.-K.V.; Humm, J.; DiResta, G.; Arbit, E.; Larson, S.; Finn, R.; Rosenblum, M.; Nguyen, H.; Gonzalez, G.; et al. Pharmacokinetics and Acute Toxicology of Intraventricular 131I-Monoclonal Antibody Targeting Disialoganglioside in Non-Human Primates. J. Neurooncol. 1997, 35, 101–112.
- Kramer, K.; Pandit-Taskar, N.; Kushner, B.H.; Zanzonico, P.; Humm, J.L.; Tomlinson, U.; Donzelli, M.; Wolden, S.L.; Haque, S.; Dunkel, I.; et al. Phase 1 Study of Intraventricular 131I-Omburtamab Targeting B7H3 (CD276)-Expressing CNS Malignancies. J. Hematol. Oncol. 2022, 15, 165.
- Pandit-Taskar, N.; Zanzonico, P.B.; Kramer, K.; Grkovski, M.; Fung, E.K.; Shi, W.; Zhang, Z.; Lyashchenko, S.K.; Fung, A.M.; Pentlow, K.S.; et al. Biodistribution and Dosimetry of Intraventricularly Administered 124 I-Omburtamab in Patients with Metastatic Leptomeningeal Tumors. J. Nucl. Med. 2019, 60, 1794–1801.
- Souweidane, M.M.; Kramer, K.; Pandit-Taskar, N.; Zhou, Z.; Haque, S.; Zanzonico, P.; Carrasquillo, J.A.; Lyashchenko, S.K.; Thakur, S.B.; Donzelli, M.; et al. Convection-Enhanced Delivery for Diffuse Intrinsic Pontine Glioma: A Single-Centre, Dose-Escalation, Phase 1 Trial. Lancet Oncol. 2018, 19, 1040–1050.
- Kramer, K.; Kushner, B.H.; Modak, S.; Pandit-Taskar, N.; Tomlinson, U.; Wolden, S.L.; Zanzonico, P.; John, H.L.; Haque, S.; Souweidane, M.M.; et al. A Curative Approach to Central Nervous System Metastases of Neuroblastoma. Pediatr. Blood Cancer 2019, 66, e27989.
- Modak, S.; Zanzonico, P.; Grkovski, M.; Slotkin, E.K.; Carrasquillo, J.A.; Lyashchenko, S.K.; Lewis, J.S.; Cheung, I.Y.; Heaton, T.; LaQuaglia, M.P.; et al. B7H3-Directed Intraperitoneal Radioimmunotherapy With Radioiodinated Omburtamab for Desmoplastic Small Round Cell Tumor and Other Peritoneal Tumors: Results of a Phase I Study. J. Clin. Oncol. 2020, 38, 4283–4291.
- Desantes, K.; Maris, J.M.; McDowell, K.; Mackall, C.; Shankar, S.; Vasselli, J.; Chen, F.; Loo, D.; Moore, P.A.; Wigginton, J.M.; et al. A Phase 1, Open-Label, Dose Escalation Study of Enoblituzumab (MGA271) in Pediatric Patients with B7-H3-Expressing Relapsed or Refractory Solid Tumors. J. Clin. Oncol. 2017, 35, TPS2596.
- Mascarenhas, L.; Chi, Y.-Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination with Chemotherapy for First Relapse Rhabdomyosarcoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866–2874.
- Ferrari, A.; Merks, J.H.M.; Chisholm, J.C.; Orbach, D.; Brennan, B.; Gallego, S.; van Noesel, M.M.; McHugh, K.; van Rijn, R.R.; Gaze, M.N.; et al. Outcomes of Metastatic Non-Rhabdomyosarcoma Soft Tissue Sarcomas (NRSTS) Treated within the BERNIE Study: A Randomised, Phase II Study Evaluating the Addition of Bevacizumab to Chemotherapy. Eur. J. Cancer 2020, 130, 72–80.
- Narayana, A.; Kunnakkat, S.; Chacko-Mathew, J.; Gardner, S.; Karajannis, M.; Raza, S.; Wisoff, J.; Weiner, H.; Harter, D.; Allen, J. Bevacizumab in Recurrent High-Grade Pediatric Gliomas. Neuro Oncol. 2010, 12, 985–990.
- Packer, R.J.; Jakacki, R.; Horn, M.; Rood, B.; Vezina, G.; MacDonald, T.; Fisher, M.J.; Cohen, B. Objective Response of Multiply Recurrent Low-Grade Gliomas to Bevacizumab and Irinotecan. Pediatr. Blood Cancer 2009, 52, 791–795.
- Moreno, L.; Moroz, V.; Owens, C.; Valteau-Couanet, D.; Gambart, M.; Castel, V.; van Eijkelenburg, N.; Castellano, A.; Nysom, K.; Gerber, N.; et al. Bevacizumab for Children with Relapsed & Refractory High-Risk Neuroblastoma (RR-HRNB): Results of the BEACON-Neuroblastoma Randomized Phase II Trial—A European ITCC-SIOPEN Trial. Ann. Oncol. 2019, 30, v901.
- Trippett, T.M.; Herzog, C.; Whitlock, J.A.; Wolff, J.; Kuttesch, J.; Bagatell, R.; Hunger, S.P.; Boklan, J.; Smith, A.A.; Arceci, R.J.; et al. Phase I and Pharmacokinetic Study of Cetuximab and Irinotecan in Children with Refractory Solid Tumors: A Study of the Pediatric Oncology Experimental Therapeutic Investigators’ Consortium. J. Clin. Oncol. 2009, 27, 5102–5108.
- Rajappa, P.; Krass, J.; Riina, H.A.; Boockvar, J.A.; Greenfield, J.P. Super-Selective Basilar Artery Infusion of Bevacizumab and Cetuximab for Multiply Recurrent Pediatric Ependymoma. Interv. Neuroradiol. 2011, 17, 459–465.
- Macy, M.E.; Kieran, M.W.; Chi, S.N.; Cohen, K.J.; MacDonald, T.J.; Smith, A.A.; Etzl, M.M.; Kuei, M.C.; Donson, A.M.; Gore, L.; et al. A Pediatric Trial of Radiation/Cetuximab Followed by Irinotecan/Cetuximab in Newly Diagnosed Diffuse Pontine Gliomas and High-Grade Astrocytomas: A Pediatric Oncology Experimental Therapeutics Investigators’ Consortium Study. Pediatr. Blood Cancer 2017, 64, e26621.
- McCrea, H.J.; Ivanidze, J.; O’Connor, A.; Hersh, E.H.; Boockvar, J.A.; Gobin, Y.P.; Knopman, J.; Greenfield, J.P. Intraarterial Delivery of Bevacizumab and Cetuximab Utilizing Blood-Brain Barrier Disruption in Children with High-Grade Glioma and Diffuse Intrinsic Pontine Glioma: Results of a Phase I Trial. J. Neurosurg. Pediatr. 2021, 28, 371–379.
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging Novel Agents for Patients with Advanced Ewing Sarcoma: A Report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force . F1000Research 2019, 8, 493.
- Gajdosik, Z. Racotumomab—A Novel Anti-Idiotype Monoclonal Antibody Vaccine for the Treatment of Cancer. Drugs Today 2014, 50, 301-7.
- Cacciavillano, W.; Sampor, C.; Venier, C.; Gabri, M.R.; de Dávila, M.T.G.; Galluzzo, M.L.; Guthmann, M.D.; Fainboim, L.; Alonso, D.F.; Chantada, G.L. A Phase I Study of the Anti-Idiotype Vaccine Racotumomab in Neuroblastoma and Other Pediatric Refractory Malignancies. Pediatr. Blood Cancer 2015, 62, 2120–2124.
- Judith Wienke; Miranda P. Dierselhuis; Godelieve A.M. Tytgat; Annette Künkele; Stefan Nierkens; Jan J. Molenaar; The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer 2020, 144, 123-150.
- Terry, R.L.; Meyran, D.; Ziegler, D.S.; Haber, M.; Ekert, P.G.; Trapani, J.A.; Neeson, P.J Immune Profiling of Pediatric Solid Tumors. J. Clin. Investig 2020, 130, 3391–3402.
- Laura R. Saunders; Alexander J. Bankovich; Wade C. Anderson; Monette A. Aujay; Sheila Bheddah; KristenAnn Black; Radhika Desai; Paul A. Escarpe; Johannes Hampl; Amy Laysang; et al.David LiuJavier Lopez-MolinaMilly MiltonAlbert ParkMarybeth A. PyszHui ShaoBrian SlingerlandMichael TorgovSamuel A. WilliamsOrit FoordPhilip HowardJacek JassemAndrzej BadzioPiotr CzapiewskiDavid H. HarpoleAfshin DowlatiPierre P. MassionWilliam D. TravisM. Catherine PietanzaJ. T. PoirierCharles M. RudinRobert A. StullScott J. Dylla A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7, 302ra136-302ra136.
- Yen Phung; Wei Gao; Yan-Gao Man; Satoshi Nagata; Mitchell Ho; High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. mAbs 2012, 4, 592-599.
- Weidle, U.H.; Eggle, D.; Klostermann, S L1-CAM as a Target for Treatment of Cancer with Monoclonal Antibodies. Anticancer. . Anticancer. Res. 2009, 29, 4919.
- Kendsersky, N.M.; Lindsay, J.; Kolb, E.A.; Smith, M.A.; Teicher, B.A.; Erickson, S.W.; Earley, E.J.; Mosse, Y.P.; Martinez, D.; Pogoriler, J.; et al.et al The B7-H3–Targeting Antibody–Drug Conjugate M276-SL-PBD Is Potently Effective Against Pediatric Cancer Preclinical Solid Tumor Models.. Clin. Cancer Res. 2021, 27, 2938-2946.
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging Novel Agents for Patients with Advanced Ewing Sarcoma: A Report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force . F1000Research 2019, 8, 493.
- Gajdosik, Z. Racotumomab—A Novel Anti-Idiotype Monoclonal Antibody Vaccine for the Treatment of Cancer. Drugs Today 2014, 50, 301-7.
- Cacciavillano, W.; Sampor, C.; Venier, C.; Gabri, M.R.; de Dávila, M.T.G.; Galluzzo, M.L.; Guthmann, M.D.; Fainboim, L.; Alonso, D.F.; Chantada, G.L. A Phase I Study of the Anti-Idiotype Vaccine Racotumomab in Neuroblastoma and Other Pediatric Refractory Malignancies. Pediatr. Blood Cancer 2015, 62, 2120–2124.
- Busch,W Aus Der Sitzung Der Medicinischen . Berl.Klin.Wochenschr. 1868, 5, 137.
- Nicholas Aps Buss; Simon J Henderson; Mary McFarlane; Jacintha M Shenton; Lolke de Haan; Monoclonal antibody therapeutics: history and future. Curr. Opin. Pharmacol. 2012, 12, 615-622.
- Köhler, G; Milestein, C Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495-497.
- Asher Mullard; FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491-495.
- Shannon L. Maude; Theodore W. Laetsch; Jochen Buechner; Susana Rives; Michael Boyer; Henrique Bittencourt; Peter Bader; Michael R. Verneris; Heather E. Stefanski; Gary D. Myers; et al.Muna QayedBarbara De MoerlooseHidefumi HiramatsuKrysta SchlisKara L. DavisPaul L. MartinEneida R. NemecekGregory A. YanikChristina PetersAndre BaruchelNicolas BoisselFrancoise MechinaudAdriana BalduzziJoerg KruegerCarl H. JuneBruce L. LevinePatricia WoodTetiana TaranMimi LeungKaren T. MuellerYiyun ZhangKapildeb SenDavid LebwohlMichael A. PulsipherStephan A. Grupp Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl. J. Med. 2018, 378, 439-448.
- Carl H. June; Roddy S. O’connor; Omkar U. Kawalekar; Saba Ghassemi; Michael C. Milone; CAR T cell immunotherapy for human cancer. Sci. 2018, 359, 1361-1365.
- M.A. Martin-Mateos; Monoclonal antibodies in Pediatrics: use in prevention and treatment. null 2007, 35, 145-150.
- Tara Gonzales; Aurore Bergamasco; Tiffany Cristarella; Camille Goyer; Matthew Wojdyla; Abiola Oladapo; John Sawicky; John Yee; Yola Moride; Effectiveness and Safety of Palivizumab for the Prevention of Serious Lower Respiratory Tract Infection Caused by Respiratory Syncytial Virus: A Systematic Review. Am. J. Perinatol. 2022, 257, 1193-2558.
- Orders, M An EUA for sotrovimab for treatment of COVID-19.. null 2021, 63, 97-xx98.
- Orders, M An EUA for casirivimab and imdevimab for COVID-19.. null 2020, 62, 201-202.
- Orders, M An EUA for bamlanivimab and etesevimab for COVID-19.. null 2021, 63, 49-50.
- Judith Wienke; Miranda P. Dierselhuis; Godelieve A.M. Tytgat; Annette Künkele; Stefan Nierkens; Jan J. Molenaar; The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer 2020, 144, 123-150.
- Terry, R.L.; Meyran, D.; Ziegler, D.S.; Haber, M.; Ekert, P.G.; Trapani, J.A.; Neeson, P.J Immune Profiling of Pediatric Solid Tumors. J. Clin. Investig 2020, 130, 3391–3402.
- Laura R. Saunders; Alexander J. Bankovich; Wade C. Anderson; Monette A. Aujay; Sheila Bheddah; KristenAnn Black; Radhika Desai; Paul A. Escarpe; Johannes Hampl; Amy Laysang; et al.David LiuJavier Lopez-MolinaMilly MiltonAlbert ParkMarybeth A. PyszHui ShaoBrian SlingerlandMichael TorgovSamuel A. WilliamsOrit FoordPhilip HowardJacek JassemAndrzej BadzioPiotr CzapiewskiDavid H. HarpoleAfshin DowlatiPierre P. MassionWilliam D. TravisM. Catherine PietanzaJ. T. PoirierCharles M. RudinRobert A. StullScott J. Dylla A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7, 302ra136-302ra136.
- Yen Phung; Wei Gao; Yan-Gao Man; Satoshi Nagata; Mitchell Ho; High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. mAbs 2012, 4, 592-599.
- Weidle, U.H.; Eggle, D.; Klostermann, S L1-CAM as a Target for Treatment of Cancer with Monoclonal Antibodies. Anticancer. . Anticancer. Res. 2009, 29, 4919.
- Kendsersky, N.M.; Lindsay, J.; Kolb, E.A.; Smith, M.A.; Teicher, B.A.; Erickson, S.W.; Earley, E.J.; Mosse, Y.P.; Martinez, D.; Pogoriler, J.; et al.et al The B7-H3–Targeting Antibody–Drug Conjugate M276-SL-PBD Is Potently Effective Against Pediatric Cancer Preclinical Solid Tumor Models.. Clin. Cancer Res. 2021, 27, 2938-2946.
- Nathaniel W. Mabe; Min Huang; Guillermo N. Dalton; Gabriela Alexe; Daniel A. Schaefer; Anna C. Geraghty; Amanda L. Robichaud; Amy S. Conway; Delan Khalid; Marius M. Mader; et al.Julia A. BelkKenneth N. RossMichal ShefferMiles H. LindeNghi LyWinnie YaoMaria Caterina RotirotiBenjamin A. H. SmithMarius WernigCarolyn R. BertozziMichelle MonjeConstantine S. MitsiadesRavindra MajetiAnsuman T. SatpathyKimberly StegmaierRobbie G. Majzner Transition to a mesenchymal state in neuroblastoma confers resistance to anti-GD2 antibody via reduced expression of ST8SIA1. Nat. Cancer 2022, 3, 976-993.
- Busch, W. A Aus Der Sitzung Der Medicinischen . Berl. Klin. Wochenschr. 1868, 13, 137.
- Busch, W Aus Der Sitzung Der Medicinischen. Berl. Klin. Wochenschr. 1868, 5, 137.