Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Most Arifa Sultana and Version 2 by Catherine Yang.
Alzheimer’s disease (AD) is a progressive disease, where dementia symptoms gradually worsen. The causes of AD are complex and are characterized by changes in the brain that lead to the accumulation of two proteins, amyloid beta and tau, forming structures called plaques and tangles, respectively. It is challenging to identify the mechanisms for the initiation and progression of AD. Oxidative stress and inflammation derived from peripheral mitochondrial dysfunction have also been suggested as alternative contributors of AD pathogenesis.
- Alzheimer’s disease
- mitochondrial dysfunctions
- metabolic disorders
1. Justification for the Mitochondrial Dysfunction Hypothesis in AD Pathogenesis
Since the discovery of the two pathological hallmarks of AD, the extracellular deposition of Aβ plaque and intracellular formation of P-tau protein have been identified as the cause of neurodegeneration of the brain. The identification of inherited mutations on the APP, presenilin 1, and 2 genes accelerate the accumulation of Aβ, leading to the development of the amyloid cascade hypothesis for AD onset. However, these mutated genes, primarily thought of as the leading causes of AD, are responsible for the occurrence of approximately 1–5% of AD in patients. The major portion of AD, >95% of the total AD population, is attributed to the Aβ plaques and P-tau. This form of AD, called sporadic AD, has been reported to be a result of apolipoprotein E4; a significant risk factor, which may accelerate Aβ formation and plaque deposition. Further establishment of the hypothesis came from the findings of multiple preclinical studies with cells and transgenic animal models showing Aβ plaque deposition as a causative pathogenesis of AD [1][2][43,58].
However, several recent studies have found that there is no significant relationship between the level or density of Aβ plaque and cognitive deficits. Moreover, the fact that drugs developed based on the amyloid cascade hypothesis are unable to treat the manifestations of dementia despite reducing Aβ further weakens the hypothesis to some extent. Evidence of late shows that individuals that died due to old age had significant Aβ deposition in the brain without showing any AD-associated symptoms during their lifetime. On the other hand, patients with or without symptoms of cognitive loss were reported to suffer from neurodegeneration even without a diagnosis of Aβ deposition [3][4][21,59]. One of the most significant findings to refute the amyloid cascade hypothesis was that treatment with a vaccine could remove Aβ plaques in AD patients, although the pathogenesis of the disease kept progressing [5][60].
In an aging brain, the normal physiological functions of mitochondria are perturbed including abnormal energy homeostasis, excessive ROS production, and mtDNA damage. Most of these abnormalities are due to dysfunctions in mitochondria, the center of ATP production and regulator of energy homeostasis. In addition, mitochondrial malfunctions lead to increased oxidative stress and decreased bioenergetics and thereby stimulate Aβ production. This finding triggers the thought of a mitochondrial cascade hypothesis, which suggests that altered mitochondrial function plays a significant role in the enhancement of Aβ production and plaque deposition [2][58]. The above evidence reasonably disproves the amyloid cascade hypothesis and indicates an alternative mechanism termed the mitochondrial cascade hypothesis in AD pathogenesis. The hypothesis infers mitochondrial dysfunction as a central cause for Aβ and tau deposition. This hypothesis, however, has been revised in recent years into primary and secondary cascades. Though the primary cascade hypothesis remains the same, the secondary cascade hypothesis theorized that the accumulation of Aβ induces mitochondrial dysfunction as an intermediate step in the disease development pathway [1][43]. This hypothesis is receiving more consideration and acceptance from the scientific community due to the emerging data from multiple scientific studies.
2. Mitochondrial Dysfunction and Alzheimer’s Disease
The majority of mitochondrial defects directly or indirectly contribute to AD pathogenesis. In other words, studies with the AD brain have revealed multiple types of mitochondrial dysfunction in the brain raising the question of whether such impairments are responsible for AD development, or if the occurrence of AD stimulates mitochondrial defects. It has been demonstrated that mitochondria can be easily accessible to Aβ, which interferes with several indigenous proteins resulting in mitochondrial dysfunction. The sources of Aβ in mitochondria might be those produced in Golgi/ER or the APP associated with mitochondria. A more recent view of mitochondrial APP metabolism has suggested the origin of Aβ in mitochondria is locally produced [6][77]. The mitochondrial production of ROS influences many metabolic processes and is usually controlled by the detoxification capacity of cellular antioxidant mechanisms. However, in the case of stress or sickness, the oxidant–antioxidant mechanism of mitochondria collapses leading to imbalanced energy production by damaging mitochondrial bioenergetic machineries [7][78]. Although the mutual causative effects of oxidative stress and the defects of mitochondria are still unclear [8][79], disruption of the balanced mitochondrial function may set off the pathogenesis of multiple disorders including AD (Figure 13). Studies show that mitochondrial ROS production leads to a vicious cycle in which an elevated level of Aβ triggers mitochondrial dysfunction along with more Aβ production eventually resulting in the advancement of sporadic AD [9][80].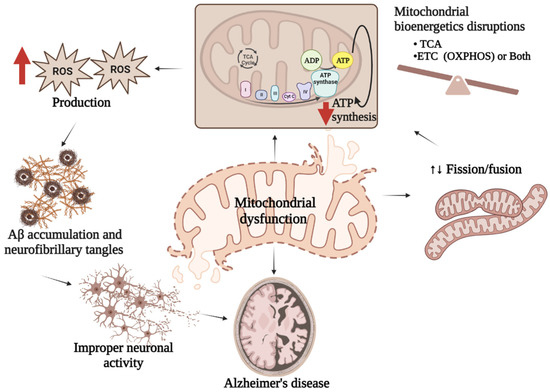
Figure 13. Mitochondrial dysfunction and AD. Mitochondrial dysfunctions characterized either by imbalanced mitochondrial bioenergetics, or impaired mitochondrial biogenesis or both result in reduced ATP synthesis followed by excess ROS generation which stimulate Aβ production leading to improper neuronal activity associated with AD pathologies.
2.1. Central Mitochondrial Dysfunction
Mitochondrial defects in the brain, so far, have been reported to be more closely related to AD than those of the peripheries. In transgenic mice, overexpression of mitochondrial Aβ-binding enzyme (ABAD) interacts with increased Aβ in the mitochondria, accelerating neuronal oxidative stress, impaired cognitive function, and memory loss [11][82]. A study with an APP transgenic mice model demonstrated that the association between APP and mitochondrial membrane obstructed the mitochondrial channels through APP/Aβ deposition on the membrane. As a result, the entry of functional molecules and enzymes was blocked, which in turn led to the accumulation of toxic substances, e.g., hydrogen peroxide [12][83]. Apart from the APP/Aβ-mediated mitochondrial disorders, the abnormal formation of Aβ is stimulated due to mitochondrial dysfunctions. A similar association of phosphorylated tau with AD neuropathology was shown in human studies using platelet-derived mitochondria demonstrating enzymatic mitochondrial dysfunction in AD progression [13][84]. Research by Yao et al. with 3xTg female mice demonstrated mitochondrial dysfunction in early stages, even before the obvious accumulation of Aβ plaques or tau tangles had occurred. Altered bioenergetic pathways were shown in the hippocampal cells derived from mouse embryos including reduced mitochondrial respiration as well as enhanced glycolysis [14][85]. This remarkable study indicated the occurrence of brain mitochondrial dysfunction at an early stage of AD pathology. From a hereditary point of view, the APOE allele is a major genetic risk factor for the initiation of sporadic AD. The impact of APOE on the accumulation of Aβ is well documented with significant evidence of Aβ clearance by APOEε2 gene therapy in the APP Tg2576 mice model [15][86]. Moreover, further studies with human brains from young individuals having APOEε4, showed reduced mitochondrial COX activity in the brain [16][87]. In the AD brain, the lipid composition of the mitochondrial cell membrane is altered resulting in a reallocation of proteins associated with the β-amyloidogenic pathway of lipid rafts [17][88]. A polyunsaturated ω-3 fatty acid, docosahexaenoic acid (DHA), was reported to pass through the BBB and contribute to the construction of the neuronal membrane. This prominent DHA could further accelerate the nonamyloidogenic processing of APP as well as reduce amyloidogenic activities of β- and γ-secretase [18][89]. Research has shown an association of toxic Aβ production and tau hyperphosphorylation with reduced FoxO activity in the brain under an insulin resistance state. This decreased expression of FoxO results from direct phosphorylation by AMPK when there is insufficient energy available [19][20][90,91]. This evidence indicates central mitochondrial dysfunction precedes AD development and progression and influences the central AD pathogenesis.2.2. Peripheral Mitochondrial Dysfunction
Apart from the effects of central mitochondrial dysfunction in AD, the contribution of peripheral mitochondrial defects has also been suggested by multiple lines of emerging data. Research with peripheral tissues such as fibroblasts in humans and mice [21][92] as well as platelets and lymphocytes demonstrated the association between mitochondrial dysfunction and altered ROS production in the periphery similar to that observed during AD in the brain. Aβ load in the brain is exacerbated through enhanced lipoprotein release in the periphery. The brain maintains its crosstalk with the periphery by lipoprotein ApoE and ApoJ with greater activity of ApoE-Aβ conjugation, resulting in reduced brain efflux of Aβ, which is accelerated when there is lower lipidation of ApoE [22][93]. Although lipids do not provide energy to the brain, they are significantly present in the mature brain, constituting the membrane structure and performing signal transduction. At the beginning of AD pathogenesis, fatty acids and acetyl-CoA produced through lipolysis and mitochondrial β-oxidation in a fed state participate in ketogenesis induced by inflammatory factors. Under energy deficit conditions, acetoacetate and β-Hydroxybutyrate (β-HB) ketone bodies serve as alternative energy sources for neurons. Generation of β-HB from acetyl-CoA, used as a substrate of acetoacetate, is also stimulated by interleukin-6 (IL-6)-induced p38/NF-κB. The entire process from lipolysis to β-HB takes place in the liver from where the β-HB crosses the BBB to provide the brain with energy under hypometabolic conditions in AD [23][20]. Moreover, the liver is stimulated by glucose metabolic dysfunction in the brain and supplies ketone bodies to the brain as an energy source in the early stages of AD [24][94], indicating a connection between the peripheral organ and the central part of the body under adverse conditions. While the brain obtains energy only from glucose, the peripheral parts can utilize both glucose and lipids as energy sources. Insulin resistance and hyperglycemia in the peripheries can stimulate a bioenergy deficit in the brain, which further deteriorates AD pathology. Central and peripheral alterations in extracellular glucose sensitize the mitochondria and result in AMPK/Akt signal fluctuations by inducing mitochondrial changes. Furthermore, studies have reported a PGC-1α-mediated reduction in mitochondria and PI3K-regulated insulin resistance in both the peripheries and the brain due to the disrupted AMPK pathway. The resulting abnormal glucose metabolism and damaged neurons can be insightful to the connection between hyperglycemia and cognitive decline [25][26][95,96].3. Role of Peripheral Mitochondrial Dysfunctions in the Initiation of AD
It is well documented that APP is expressed both in neuronal cells and in peripheral tissues such as the heart, liver, pancreas, blood cells, and kidneys [25][95]. Moreover, healthy individuals have lower levels of Aβ in the brain and peripheral tissues than that of cognitively impaired individuals. Though there are differences in central and peripheral APP processing and Aβ isoform deposition, there is reduced Aβ in the periphery [27][28][29][97,98,99] and there is evidence of Aβ interconnection between the brain and the periphery. Aβ originated in the brain can be cleared from the brain through several pathways; one of which is the transportation or the efflux of Aβ across the BBB to the peripheral circulation. Apart from the efflux, there could be an influx of Aβ from the periphery to the brain. The potential role of the efflux and the influx might balance the Aβ pool between the brain and periphery. The peripheral mitochondrial origin of Aβ may have systemic effects and stimulate CNS damage after crossing BBB. Several studies including humans and mice have demonstrated the occurrence of Aβ accumulation in the brain when induced peripherally with Aβ extracted from the brain, suggesting its systemic circulation between the periphery and the brain [30][62]. Studies with APP transgenic mice to test the effects of intraperitoneal injections of Aβ containing brain extract have shown robust cerebral beta-amyloidosis in all the inoculated mice [31][100]. Further, human studies with subcutaneous Aβ inoculation revealed seeding effects of Aβ, which can spread to the CNS after injection and seed in the parenchyma of the brain [32][101]. The interaction of Aβ with RAGE in human brain endothelial cells facilitates the transportation of Aβ across the BBB, resulting in the release of proinflammatory cytokines in the brain. Higher expression of RAGE was also reported in the AD brain compared with age-matched controls [33][102]. These data legitimately suggest the transfer of Aβ produced in the periphery to the brain with a high potentiality of triggering clinical manifestations of AD. Oxidative stress due to mitochondrial dysfunction has been proposed as a common pathological mechanism underlying most of the chronic neurodegenerative diseases including AD. Whereas inflammation is a body’s protective response against multiple insults, uncontrolled inflammation can result in enormous cell and tissue damage. Due to its vulnerability, the BBB can be highly affected by peripheral inflammation. Under diseased conditions, peripheral immune cells such as macrophages cross the BBB, altering the CNS environment and accelerating chronic neurodegeneration. Further, microglia and astrocytes become activated upon disruptions in the CNS triggering the secretion of proinflammatory cytokines. Moreover, ROS production by activated microglia and astrocytes can be responsible for neuronal damage [34][103]. Studies with APP transgenic mice have shown increased BBB permeability following peripheral injection of inflammatory substances such as lipopolysaccharide (LPS) facilitating the accumulation of peripheral proinflammatory molecules including TNF-α, and IL-6. Such infiltration of cytokines stimulates neuronal inflammation and subsequent disease progression [35][71]. Therefore, the evidence of the systemic circulation of Aβ and the impacts of peripheral oxidative stress as well as inflammation on CNS upon crossing the BBB provides potential involvement of the consequences of peripheral mitochondrial dysfunction in the onset and progression of AD. Based on the discussion, the article summarizes (Figure 24) possible linkages among metabolic disorders, mitochondrial dysfunction, and AD for visualization. There is evidence that peripheral metabolic impairments can influence peripheral mitochondrial dysfunctions. On the other hand, mitochondrial dysfunctions can be the consequence of altered metabolic functions. Therefore, the interrelationship between metabolic disorders, mitochondrial dysfunction, and AD might involve multiple complex and likely common pathways.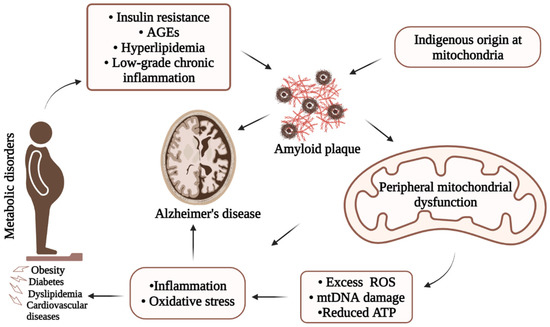
Figure 24. Simplified illustration of underlying AD pathology showing possible interrelation among metabolic disorders, mitochondrial dysfunction, and Alzheimer’s disease. Due to its complexity, onset of AD involves several major pathways including metabolic disorders and mitochondrial dysfunctions through the production of Aβ, increased inflammatory responses, and/or oxidative stresses.