Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Amanda Jan Unsworth and Version 4 by Wendy Huang.
Pim Kinases; Pim-1, Pim-2, and Pim-3, are a family of constitutively active serine/threonine kinases, widely associated with cell survival, proliferation, and migration. Historically considered to be functionally redundant, independent roles for the individual isoforms have been described. Whilst most established for their role in cancer progression, there is also increasing evidence for wider pathological roles of Pim kinases within the context of cardiovascular disease, including inflammation, thrombosis, and cardiac injury.
- Pim kinase
- Pim-1
- Pim-2
- Pim-3
- microRNA
1. Introduction
Proviral integration site for MuLV (Murine Leukaemia Virus) (Pim) Kinases are a family of serine/threonine kinases located in numerous cell types throughout the body [1][4]. Within the Pim kinase family, there are three isoforms, Pim-1, Pim-2, and Pim-3, and their roles include promoting cell survival [2][3][4][5][5,6,7,8], proliferation [6][9], and migration [7][10]. They are implicated in the pathogenesis of various diseases, with roles identified in multiple cell types, but are most widely known for their contribution to haematological and solid tumour malignancies, which have been reviewed elsewhere [8][9][10][11][11,12,13,14]. As Pim kinases are constitutively active [12][13][15,16], they have become a potential target for cancer therapy, with numerous inhibitors being developed to treat myeloma and myelofibrosis, in addition to other malignancies [14][15][16][17,18,19]. Interestingly, their expression and activity are not limited to cancer, with the Pim kinase isoforms demonstrating wider pathological roles of Pim kinases including including inflammation, thrombosis, and cardiac injury.
2. Structure, Regulation, and Localisation of Pim Kinases
Structure, Regulation, and Localisation of Pim Kinases
Whilst the three Pim kinase family members are named isoforms due to being highly conserved with high amino acid sequence homology (Figure 1), they are encoded by separate genes and located on different chromosomes (Figure 2). They are classed as serine/threonine kinases, yet the Pim kinases uniquely lack regulatory domains, with only kinase domains present [17][22]. The kinases do adopt the commonly observed bi-lobed kinase fold structure seen in other kinases; however, the Pim family members contain a novel two-stranded β-sheet predating the αc helix, which distinguishes them from other kinases [18][23]. Another distinctive feature of Pim kinases is that they contain a unique hinge region [12][18][15,23], which makes targeting the kinase family attractive. The hinge region of Pim kinases has two inserted proline residues, widening the ATP (adenosine triphosphate) binding site. This widened ATP site does not allow for the typical hydrogen bonds between ATP and kinases and results in high Km (Michaelis constant) values for ATP [19][24].

Figure 1. Sequence alignment of Human Pim family kinases. Sequences were aligned using Clustal Omega [20][25] and ESPript 3 [21][26] using Uniprot accession sequences Pim-1—P11309, Pim-2—Q9P1W9, and Pim-3—Q86V86. Exact matches are shown in white text on a red background, and similar residues in boxed yellow and black text on a white background are little consensus between the three isoforms.
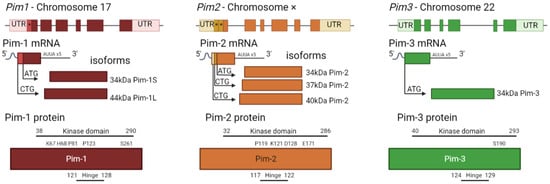
Figure 2. Pim kinase family gene structures and corresponding protein structures. Each gene has six codons surrounded by large untranslated regions (UTR). Alternative start codons within the gene (*) correspond to alternate mRNA transcripts, representing the different isoforms. Each isoform consists of only a serine/threonine kinase domain, with molecular weights varying from 34–44 kDa. The largest isoform is shown in more detail as to where the kinase domain and hinge regions are present and where key sites are located (not to scale). Pim-1 key sites include K67, H68 (site mutation increases kinase activity [18][23]), P81 (site mutation decreases kinase activity [18][23]), P123 (proline region in hinge site which is unique to Pim family kinases [8][12][11,15]), and S261 (phosphorylation site [22][27]). Pim-2 key sites include P119 (proline residue in hinge site, unique to Pim kinases (Q9P1W9—[23][28]), K121 (site mutation renders kinase inactive [13][16]), D128 and E171 (required for potent drug inhibition [24][29]). Pim 3 sites are the hinge region (Q86V86 [23][28]) and S190 (predicted autophosphorylation site [25][30]).
Due to the lack of regulatory domains within the protein (Figure 2) and its protein structure, the Pim kinases are hypothesised to be constitutively active [13][26][16,31], and regulation of the kinases is via transcriptional, post-transcriptional, translational, and post-translational mechanisms (Figure 3). Transcriptional regulation of Pim kinases is usually in response to mitogenic stimulants such as platelet-derived growth factor (PDGF) in vascular smooth muscle cells [27][28][32,33], high glucose [29][34], hypoxia, [30][31][35,36] and inflammation such as in response to cytokines, IL-6 (interleukin-6) [32][37], and TNF-α (tumour necrosis factor—alpha) treatment [33][38]. Furthermore, transcription of Pim-1 has been demonstrated to be regulated downstream of the Janus kinase/signal transducer and activator of the transcription (JAK/STAT) signalling pathway, with activation of the pathway being responsible for its upregulation [34][39]. Examples of this can be observed downstream of both the erythropoietin (EPO) receptor and thrombopoietin (TPO) receptor (c-Mpl) [35][40], where EPO and TPO can induce the expression of Pim-1 via JAK/STAT signalling [36][37][41,42].
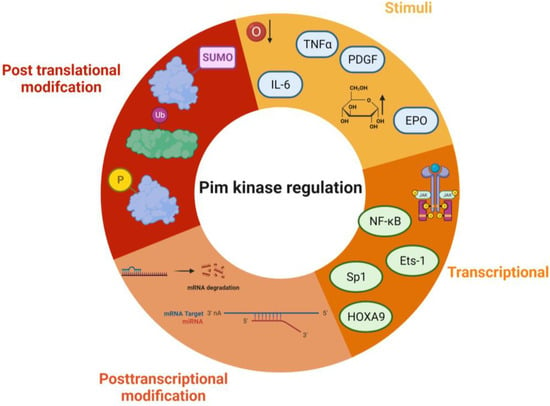
Figure 3. Regulation of Pim kinases. Some of the key regulatory pathways that control Pim kinase activity and signalling. Stimuli—TNFα—Tumour necrosis factor alpha, EPO—erythropoietin, IL-6—interleukin 6, PDGF—platelet-derived growth factor, hypoxia, and high glucose. Transcriptional modifications include NF-Kb—Nuclear factor kappa B, Ets-1—ETS proto-oncogene 1, Sp1—Specificity Protein 1, and HOXA9—Homeobox A9. Post-translational modifications include ubiquitination, sumoylation, and phosphorylation. Post-transcriptional modification—mRNA degradation, and miRNA binding. The figure was created using Biorender.
Post-transcriptional modifications are related to mRNA processing. Within the mRNA sequence of each isoform are five AUUA destabilising motifs in the untranslated region (UTR) at 3′ [38][39][43,44] (Figure 34), which target the transcripts for rapid degradation [40][45]. Indeed, this appears to be a key mechanism for the regulation of the Pim kinases, with the half-life of Pim-1 mRNA being approximately 25 min in vascular smooth muscle cells [28][33].
However, this may not be the sole mechanism of transcriptional regulation of Pim kinase isoform expression levels. In vascular smooth muscle cells (vSMCs), there appear to be other transcriptional methods of control, such as delayed transcription and translation. Cell stimulation with PDGFbb leads to a transient increase in Pim-1 mRNA at 1 h; however, the protein expression profile does not match this time course with protein levels peaking much later at 24 h [28][33]. In contrast, in response to high glucose, vSMC Pim-1 mRNA and protein levels do correspond to each other, with an increase in both observed at 48 h, suggesting that there may be alternative mechanisms of Pim protein translation depending upon the cellular stimuli [29][34].
Post-translational modifications of Pim kinase include phosphorylation, dephosphorylation, ubiquitination, and sumoylation. Initially considered to regulate Pim kinase activity, phosphorylation of the Pim-1 isoform on its N terminus [26][31] was demonstrated instead to control protein stability and degradation rather than catalytic activity. Various candidates have been identified as possible mediators of Pim phosphorylation including Protein Kinase C α (PKCα) and endothelial/epithelial tyrosine kinase (Etk) [41][53]. PKCα-dependent phosphorylation of Pim-1 has been demonstrated to enhance protein stability, increasing the half-life of the protein [42][54]. Unphosphorylated Pim-1 is still maintained within the active conformation, with active site residues of the kinase prepared to initiate phosphate transfer [12][26][15,31].
Pim kinase autophosphorylation is also believed to be important for kinase stability. Pim-1 was initially thought to be auto phosphorylated on serine 261; however, it has been identified that this occurs on serine 8 [26][31]. Pim-1 mutants that are unable to autophosphorylate due to mutations within their kinase structure have significantly shorter half-lives, although this could be mediated due to increased clearance [43][55].
Dephosphorylation and subsequent degradation of Pim kinase occur via protein phosphatase 2A (PP2A), with inhibition of PP2A activity increasing the half-life of Pim kinases [44][56]. However, it is not clear whether this mechanism is direct or mediated through Suppressors of Cytokine Signalling-1 (SOCS-1). SOCS-1 is involved in the degradation of proteins through ubiquitination [45][57]. Indeed, SOCS-1 binds to Pim kinases [46][58], and is required for the PP2A-mediated degradation of Pim kinase [44][56].
Pim-1 has been shown to be protected from ubiquitin-mediated proteasome degradation following heat shock via a mechanism that involves heat shock protein 90 (HSP90) binding. During the treatment of cells with geldanamycin, an HSP90 inhibitor, rapid degradation and reduced kinase activity of Pim-1 occurs [47][48][59,60]. However, ubiquitin-mediated degradation may not extend to all Pim isoforms, as ubiquitination of Pim-2 does not appear to target the kinase for degradation. Adam et al. demonstrate that inhibiting the formation of the cullin RING ligase-containing ubiquitin ligase complex via inhibition of NEDD8-Activating Enzyme did not lead to a modulation of Pim-2 protein levels, despite control proteins being altered. Furthermore, the E1-ubiquitin ligase inhibitor PYR-41 did not alter the stability of the Pim-2 protein [49][61]. In contrast, a recent study has proposed that ubiquitination of both Pim-1 and Pim-2 is important in regulating the stability of the protein, when under conditions of hypoxia, during hypoxia, deubiquitinase USP28 preferentially binds to Pim-1 and -2 and prevents their degradation [50][62], suggesting that alternative regulatory mechanisms of Pim kinase degradation may take place under hypoxic conditions.
Degradation of Pim kinases may also occur via Small Ubiquitin-like Modifier (SUMOlyation). Mutation of the potential SUMO site, E171, in Pim-1, increases the protein half-life to almost double that of wild type [43][55]. Whilst only Pim-1 was studied, Pim-2 and Pim-3 also share similar consensus sequences for the SUMOylation [43][55]. Furthermore, silencing of the SUMO-targeted E3 ubiquitin ligase RNF4 also increases Pim-1 levels. This suggests ubiquitin-facilitated proteasomal regulation of Pim kinase levels [43][55].
3. Pim-1
Of the three Pim kinase isoforms, Pim-1 is the most extensively researched family member. The Pim-1 gene is located on chromosome 6, and in-frame initiation translation codons on the chromosome result in two Pim-1 isoforms of different sizes [51][63]. Pim-1L (44 kDa) is an N-terminal extension of Pim-1S (34 kDa), initiated by an upstream CUG codon [39][52][44,64], as shown in Figure 23. Both isoforms have comparable kinase activity [39][44], but due to the extra proline-rich motif in Pim-1L, it is suggested that Pim-1L primarily localises at the plasma membrane, with Pim-1S being primarily nuclear or cytosolic [53][65]. There is little literature focusing on the differing isoforms of Pim-1; however, one study has identified that there may be subtle differences between their phosphorylation activities within the context of cancer [54][66]. Pim-1L and Pim-1S can phosphorylate the androgen receptor at serine-213, whereas only the larger isoform could phosphorylate at threonine 850 in vivo suggesting some differential phosphorylation, which may be related to their localisation within the cell [54][66]. Pim-1 expression is widely associated with hematopoietic neoplasia and solid tumours [8][17][11,22], and increased Pim-1 expression is typically a marker of poor prognosis [55][67]. Pim-1 inactivates pro-apoptotic protein BAD by phosphorylating it on Ser112 [2][5]. Furthermore, it promotes cell cycle progression through phosphorylation and activation of Cdc25 [56][57][68,69], and phosphorylation and inhibition of Cdc25-associated kinase 1 (C-TAK1) [58][70], an inhibitor of Cdc25C. The promotion of tumorigenesis through phosphorylation of c-Myc [59][60][71,72] and regulation of mTORC [61][73] is also be mediated via Pim-1. Pim-1 has also been shown to promote cell migration and chemotaxis (metastasis) via the phosphorylation of CXCR4 [62][74].4. Pim-2
Pim-2, located on the X chromosome, is the most divergent member of the kinase family (Figure 1). Similar to Pim-1, Pim-2 also has multiple isoforms, 34, 37, and 40 kDa (Figure 2) [63][75]; however, it only shares 66% amino acid sequence homology with Pim-1, with the most substantial differences being within the C terminal (23 residues) (Figure 1). Within this sequence are six proline residues that are not predicted to form a helical structure; therefore, Pim-2 lacks a C-terminal α helix [19][24] compared to the other isoforms. This lack of helical structure is thought to increase the flexibility of the terminus, resulting in structural changes to Pim-2 compared with Pim-1 and -3, which may offer an explanation as to why pan-Pim kinase inhibitors appear to be less potent on Pim-2 compared to the other Pim family members [64][65][76,77]. Modelling approaches have demonstrated that amino acids D128 and E171 are required for potent inhibition of Pim-2 but not Pim-1 and -3 [24][29]. Despite these apparent differences in structure, Pim-2, like Pim-1, can also phosphorylate c-Myc [60][72] and maintain mTORC signalling by maintaining inhibitory phosphorylation on eukaryotic initiation factor 4E binding protein (4E-BP1) and BAD, allowing for cell growth and survival [13][16]. All three Pim-2 isoforms are cytoplasmic; however, they do have different levels of expression and protein stability. 37 kDa and 34 kDa isoforms of Pim-2 have very short half-lives (less than 30 min), whereas the 40 kDa isoform has a half-life approximately twice that of the shorter isoforms [49][61]. These differences in protein stability are likely due to the disordered N-terminal domains that can be recognised directly by the proteasome [49][61]. Differences in the activity of the Pim-2 isoforms have also been observed in vitro. The largest isoform has been shown to be less active when compared to the smaller isoforms; however, it is unclear why this is; one hypothesis is that the larger 40 kDa isoform may contain an auto-inhibitory sequence [4][7].5. Pim-3
Pim-3 is located on chromosome 22 and shares 77% sequence homology and similar protein structure with Pim-1. However, unlike the other family members, which are expressed as multiple-length variants, Pim-3 is only expressed as one 34 kDa isoform (Figure 23) [66][78]. Similar to Pim-1 and Pim-2, Pim-3 can phosphorylate BAD at Ser112, with Pim-3 knockdown shown to reduce phosphorylation at this site [5][67][8,79]. MYC activity can also be augmented by Pim-3, along with control of protein synthesis [68][80]. The summary of the key differences between the different members of the Pim kinase family can be found in Table 1.Table 1.
Comparison of the Pim kinase family members.
| Pim-1 | Pim-2 | Pim-3 | |
|---|---|---|---|
| Isoforms | 44 kDa 34 kDa [37][52][42,64] |
34 kDa 37 kDa 40 kDa [4][49][61][7,61,73] |
34 kDa [66][78] |
| Isoform differences | Preferential of targets as opposed to others for Pim-1L and Pim-1S [54][66] such as Pim-1S favouring the androgen receptor [69][81] and Pim-1L phosphorylating Etk [53][65] | Largest isoform less active [4][7[49],61] | N/A |
| Protein localisation | Pim-1L plasma membrane Pim-1S cytosolic [53][65] |
Cytoplasmic [49][61] | Cytoplasm [70][82] |
| Chromosome [71][83] | 6 [51][63] | X [71][83] | 22 [71][83] |
| Sequence similarity [19][66][24,78] | Pim-1 Pim-2 66%, Pim-1 Pim-3 77% [70][82], Pim-2 Pim-3 44% (Figure 12) |
||
| Structural differences | C-terminal helix missing in Pim-2 Six proline residues in the last structure [19][24]. |
||
| Transcriptional regulation differences |
NF-KappaB [72][84]. HOX9A upregulates Pim-1 [73][85] |
Not dependent on NF-KappaB, but Sp1 and Ets-1 [74][86]. HOX9A downregulates Pim-3 [75][87]. |
|
| Degradation | Ubiquitination [47][48][50][59,60,62] | Ubiquitin independent in normoxia [49][61]. Under hypoxia, ubiquitination takes place [50][62] | Believed to be Ubiquitinated similarly to Pim-1 [50][62] |
| Consensus peptide sequence (AKRRRRHPSGPPTA) binding affinity [26][31] |
40–60 nM | 640 nmol/L | 40–60 nM |
| Kd for consensus sequence [26][31] | 0.058 µM | 0.64 µM | 0.039 µM |