Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jessie Wu and Version 2 by Jessie Wu.
Non-small cell lung cancer (NSCLC) is a leading cause of death. Capmatinib is a Type Ib MET TKI first discovered in 2011 and was FDA approved in August 2022 for advanced NSCLC with MET exon 14 skipping mutation. Clinical trials now involve combination therapy with capmatinib, including amivantamab, trametinib, and immunotherapy. Furthermore, new drug agents, particularly antibody–drug conjugates, are being developed to help treat patients with acquired resistance from capmatinib and other tyrosine kinase inhibitors (TKIs).
- dysregulation
- capmatinib
- tyrosine kinase inhibitor
- detection
- NSCLC
1. Introduction
Non-small cell lung cancer (NSCLC) is a leading cause of death, accounting for an estimated 1.8 million deaths according to GLOBOCAN in 2020 [1]. Over the past decade, there has been tremendous progress in the discovery and development of targeted therapies for EGFR; KRAS G12C; BRAF V600E mutations; ALK, ROS1; RET gene rearrangements; MET alterations, including MET exon 14 skipping mutations, ERBB2 (HER2) mutations, and NTRK 1/2/3 gene mutations [2][3][4][5][6][7][8][9][10]. This has led to the personalization of medicine in NSCLC.
The mesenchymal–epithelial transition factor (MET) gene is located in human chromosome 7 (7q21–q31), comprising 21 exons and 21 introns, and encodes a protein that is approximately 120 kDa in size. The ligand for MET is hepatocyte growth factor (HGF), which is a soluble cytokine and is synthesized by mesenchymal cells, fibroblasts, and smooth muscle cells [11]. HGF will bind to MET, and this will trigger the autophosphorylation of Tyr-1234 and Tyr-1235 in the intracellular tyrosine kinase domain, which then undergoes further autophosphorylation of Tyr-1340 and Tyr-1356 in the C-terminal docking site [11][12]. This then facilitates the recruitment of intracellular effector molecules such as GRB2, SRC, PIK3, and GAB1, leading to the activation of downstream pathways. Normally, MET/HGF signaling pathway mediates embryogenesis, tissue regeneration, wound healing, and the formation of nerves and muscles [11][12][13].
In cancer, the MET proto-oncogene is abnormally activated and stimulates other signaling pathways in tumor cells, notably PI3K/AKT, JAK/STAT, Ras/MAPK, SRC, and Wnt/beta-catenin [11] (Figure 1). MET overexpression can be found in inflammation and hypoxia, leading to proliferation and migration, and is seen in a large variety of cancer types, including epithelial, mesenchymal, and hematological malignancies [14]. In NSCLC, it has been shown to be overexpressed in 35–72% of cases [14]. High levels of MET expression have been found to correlate with early disease recurrence [15]. MET dysregulation in NSCLC can present in a variety of ways—gene overexpression; HGF expression that can cause ligand-induced activation, leading to sustained or altered signaling; gene amplification, which can lead to overexpression and reduce the requirement for ligand activation, leading to sustained or altered signaling of the MET receptor; gene rearrangement, which may reduce or remove the requirement for ligand activation, leading to sustained altered signaling properties of the MET receptor; and downstream MET signaling alterations [11][12][15]. Notably, cigarette smoking can upregulate c-MET and the downstream Akt pathway [16]. It also affects the sensitivity of EGFR TKIs as cigarette smoke attenuates the AMP-activated protein-kinase (AMPK)-dependent inhibition of mTOR which then decreases the sensitivity of NSCLC cells with wild-type EGFR to TKI and thereby represses the expression of liver kinase B1 (LKB1) [17]. Finally, MET dysregulation can occur via gene mutation, most notably the MET exon 14 skipping mutation seen in about 3–4% of adenocarcinoma and 2% of squamous cell carcinoma but in higher frequencies in adenosquamous carcinoma (6%) and pulmonary sarcomatoid carcinoma (9–22%) [15][18].
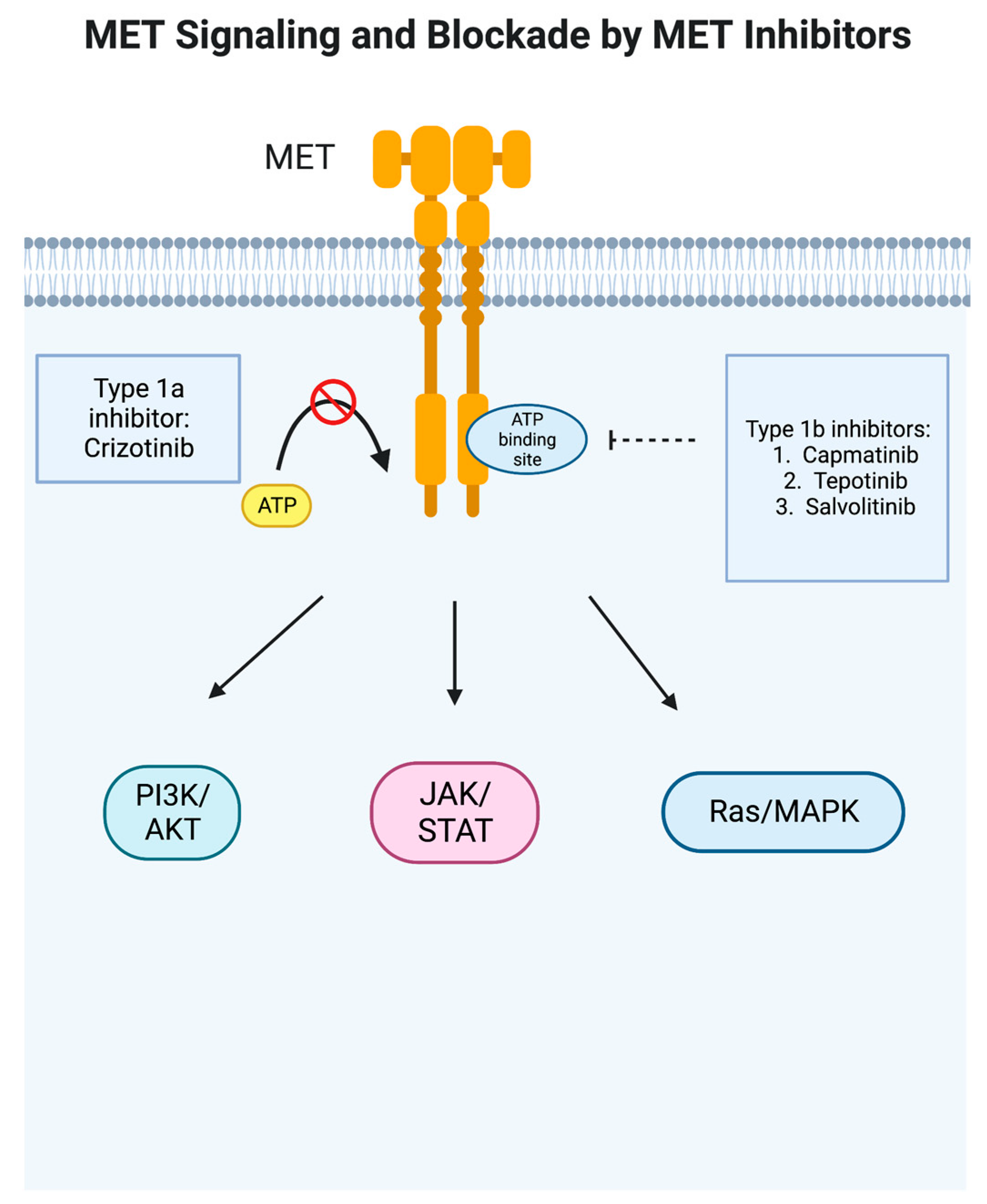
Figure 1. MET signaling pathway and blockade by MET inhibitors. In cancer, the MET proto-oncogene is abnormally activated and stimulates other signaling pathways in tumor cells, notably PI3K/AKT, JAK/STAT, Ras/MAPK, SRC, and Wnt/beta-catenin [11]. Type 1a inhibitor crizotinib blocks ATP binding to prevent the phosphorylation of the receptor, whereas type 1b inhibitors such as capmatinib are more specific and bind to a pocket adjacent to the ATP binding site. This figure was generated by BioRender.
MET exon 14 skipping mutations are processes in which the 47-amino-acid juxtamembrane domain is deleted, altered, or disrupted by intronic regions surrounding exon 14, leading to fusion in mature mRNA between exon 13 and exon 15 [19][20]. MET exon 14 skipping mutations have been shown to be exclusive from other driver mutations but coexist with other MET amplification or copy number gains [21]. Meanwhile, the amplification of the MET gene, which is defined as a gain in copy number (GCN), has been seen both de novo and as an acquired resistance mechanism [22]. MET amplification is seen in EGFR-acquired resistance and can occur with or without the loss of T790M [23]. In the analysis of resistance mechanisms in the AURA 3 study (n = 78), MET amplification was seen in (14/78,18%) of samples, EGFR C797S (14/78,18%) of cases, and 15 patients having >1 resistance-related genomic alteration [23][24]. MET amplification is also considered an acquired resistance mechanism of ALK inhibitors, as MET amplification has been observed in about 15% of next-generation ALK inhibitor resistance [25]. Both MET exon 14 skipping mutations and MET high-level amplification have been shown to portend poor prognosis [21]. Without the use of MET inhibitors, a retrospective study by Awad et al. showed that the median OS was 8.1 months [26]. MET exon 14 skipping mutations are seen more frequently in females than in males, and the median age of MET exon 14 skip mutation patients ranged from 71.4 to 76.7 years [6][18]. Compared with other driver mutations, MET exon 14 skip mutation patients tend to be smokers, with only about 36% being never smokers in a previous retrospective analysis [27].
MET tyrosine kinase inhibitors have been developed to treat MET-dysregulated NSCLC, classified as Type I, Type II, and Type III inhibitors. Type I inhibitors compete with ATP for the binding of the ATP-binding pocket of the active conformation of MET. Specifically, Type Ia inhibitors such as crizotinib interact with the Y1230 residue in the hinge region and are dependent on binding with the G1163 residue [28][29]. Type Ib inhibitors such as capmatinib, tepotinib, and savolitinib also connect with the Y1230 residue but are not dependent on G1163 binding [28][30][31][32]. Meanwhile, Type II inhibitors, which include cabozantinib, meresitinib, and gleasatanib, bind the ATP pocket in an inactive state [32][33][34][35]. Type III inhibitors bind to allosteric sites different from the ATP site and are not competitive; tivantinib has been studied in NSCLC but was not found to show any benefit in interim analysis and therefore was discontinued [32][34][36].
2. Pharmacodynamics/Pharmacokinetics
Capmatinib is a selective Type Ib ATP-competitive tyrosine kinase inhibitor targeting MET. Capmatinib has an average IC50 value of 0.13 nM and a cell-based IC50 of 0.3–0.7 nM in lung cancer cell lines [28][37] (Figure 2). Capmatinib has linear pharmacokinetics, with exposure increasing approximately dose-proportionally over a dose range of 200–400 mg. It is rapidly absorbed, with peak plasma concentration (Cmax) obtained about 1–2 h after a 400 mg dose is given. There is similar absorption when taken with and without food. The effective elimination half-life is 6.5 h. The plasma protein binding is 96% [38][39].
Figure 2. Chemical structure of capmatinib; the asterisk (*) represents the chiral carbons that are part of the chemical structure.. The chemical name for capmatinib is 2-Fluoro-N-methyl-4-[7-(quinolin-6-ylmethyl)imidazo[1,2 b][1,2,4]triazin-2-yl]benzamide—hydrogen chloride—water (1/2/1). The molecular formula for capmatinib hydrochloride is C23H21Cl2FN6O2 [38].
3. GEOMETRY Mono-1 Trial
The GEOMETRY mono-1 trial was a multicohort Phase II study in patients with MET-dysregulated advanced NSCLC. The patients were either in Stage IIIB or IV NSCLC, had no EGFR mutation, and were negative for ALK rearrangement. All subjects took capmatinib 400 mg po b.i.d. A total of 364 patients were enrolled, with 97 having a MET exon 14 skipping mutation and 210 having MET amplification. There were seven cohorts to the study: In previously treated patients (1–2 lines of therapy), Cohort 1 consisted of MET amplification with (a) GCN ≥ 10 (n = 69) or (b) GCN 6–9 (n = 42); Cohort 2 consisted of MET amplification with GCN 4–5 (n = 54); Cohort 3 consisted of MET amplification with GCN < 4 (n = 30); Cohort 4 consisted of MET exon 14 skipping mutation with any GCN (n = 69); and Cohort 6 consisted of MET amplification with GCN > 10 (n = 3) or MET exon 14 skipping mutation with any GCN (n = 31) who had received one line of therapy (n = 34). In the untreated group, Cohort 5a consisted of MET amplification with GCN ≥ 10 (n = 15); Cohort 5b consisted of MET exon 14 skipping mutation with any GCN (n = 28); and Cohort 7 consisted of treatment-naïve MET exon 14 skipping mutation with any GCN (n = 23). MET exon 14 skipping mutation patients had a slightly higher median age (71 years) than patients with MET amplification (60–70 years) on diagnosis. Patients with MET exon 14 skipping mutation were more likely to be women and to have never smoked [6]. Among patients with MET exon 14 skip mutations, ORR was seen in 41% (95% CI 29–53) of 69 previously treated patients and 68% (95% CI 48–84) of 28 previously untreated patients. The median duration of response (DOR) was 9.7 months (95% CI 5.6–13.0) among the treated patients and 12.6 months (95% CI 5.6—not reached) in previously untreated patients. Most patients (82% in treated and 68% in untreated) had a response at the first tumor evaluation following the start of capmatinib therapy. The median PFS was 5.4 months (95% CI 4.2–7.0) in previously treated patients and 12.4 months (95% CI 8.2—not reached) in previously untreated patients. Notably, 12 of 13 patients with exon 14 skipping mutations who had brain metastasis had intracranial disease control. The primary reason for discontinuation was progressive disease (58% in previously treated patients and 46% in untreated patients) [6]. In patients with GCN < 10, the cohorts were closed due to futility, as PFS for GCN 6–9 and 4 or 5 was only 2.7 months. In GCN ≥ 10, there was activity; the ORR was 29% (95% CI 19–41) in previously treated patients and 40% (95% CI 16–68) in previously untreated patients, but this fell below the predefined clinical efficacy. The median DOR was 8.3 months (95% CI 4.2–15.4) in treated patients and 7.5 months (95% CI 2.6–14.3) in untreated patients. The median PFS was 4.1 months (95% CI 2.9–4.8) in treated patients and 4.2 months (95% CI 1.4–6.9) in untreated patients [6] (Table 1).Table 1. Responses to capmatinib treatment relative to the cohort in GEOMETRY mono-1 trial (6).
| Response | NSCLC with MET Exon 14 Skipping Mutation | NSCLC with MET Amplification | |||||
|---|---|---|---|---|---|---|---|
| Best Response—No (%) | Cohort 4 n = 69, any GCN with 1–2 Lines of Therapy |
Cohort 5b n = 28, any GCN with No Previous Therapy | Cohort 1a n = 69, GCN ≥ 10 with 1–2 Lines of Therapy | Cohort 5a n = 15, GCN ≥ 10 with No Previous Therapy | Cohort 1b n = 42, GCN 6–9 with 1–2 Lines of Therapy | Cohort 2 n = 54, GCN 4 or 5 with 1–2 Lines of Therapy | Cohort 3 n = 30, GCN < 4 with 1–2 Lines of Therapy |
| Complete response | 0 | 1 (4) | 1 (1) | 0 | 0 | 0 | 0 |
| Partial Response | 28 (41) | 18 (64) | 19 (28) | 6 (40) | 5 (12) | 5 (9) | 2 (7) |
| Stable disease | 25 (36) | 7 (25) | 28 (41) | 4 (27) | 17 (40) | 20 (37) | 14 (47) |
Table 2. Adverse events in all cohorts (n = 364) in the GEOMETRY mono-1 trial [6].
| Adverse Event | Total | Grade 3 or 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Incomplete response or nonprogressive disease | ||||||||||
| Progression or death—No. of patients | ||||||||||
| 60 | ||||||||||
| Any event—No. (%) | 355 (98) | 244 (67) | ||||||||
| 17 | ||||||||||
| Most common events—No. (%) | ||||||||||
| 1 (1) | ||||||||||
| Peripheral edema | 186 (51) | 33 (9) | 1 (4) | 1 (1) | 0 | |||||
| Nausea | 1 (2) | 0 | 0 | |||||||
| 163 (45) | 9 (2) | Unknown or could not be evaluated | 9 (13) | |||||||
| Vomiting | 102 (28) | 9 (2) | 28 | 19 | 20 | 6 | ||||
| Blood creatinine increased | 89 (24) | 0 | 5 | 5 | 2 | |||||
| Dyspnea | 84 (23) | 24 (7) | Percent of patients (95% CI) | 41 (29–53) | 68 (48–84) | 29 (19–41) | 40 (16–68) | 12 (4–26) | 9 (3–20) | 7 (1–22) |
| Fatigue | 80 (22) | 16 (4) | Disease control | |||||||
| 2 (1) | ||||||||||
| Back Pain | 54 (15) | 3 (1) | ||||||||
| Pyrexia | 50 (14) | 3 (1) | ||||||||
| ALT increased | 48 (13) | 23 (6) | ||||||||
| Asthenia | 42 (12) | 13 (4) | ||||||||
| Pneumonia | 39 (11) | 17 (5) | ||||||||
| Weight loss | 36 (10) | 2 (1) | ||||||||
4. Future Directions/Conclusions
Capmatinib, a Type Ib MET TKI that is not dependent on G1163, as crizotinib is, has proven to have efficacy, as shown in the GEOMETRY mono-1 study. Subsequent post hoc analyses have shown similar efficacy regardless of the prior treatment used and patient-reported improvement in quality of life. In addition, real-world analysis has shown similar efficacy with a promising intracranial response. The Foundation One CDx assay has been shown to be a reliable companion assay and remains the only FDA-approved assay for MET-targeted therapies. However, there have been no completed Phase III studies comparing capmatinib to first-line chemotherapy and immunotherapy or second-line chemotherapy. Furthermore, there was a notable percentage of Grade 3–4 toxicities. Future studies include investigations of capmatinib with MEK inhibition, combination therapy with amivantamab, and new classes of drugs, particularly ADCs. (Table 3) Capmatinib’s role in a perioperative setting in early-stage NSCLC may provide further treatment options for early stage patients with MET exon 14 skipping NSCLC, but the sequencing of these drugs and tolerability will be key factors, along with finding a more reliable biomarker.Table 3. Current key ongoing studies involving capmatinib.
| Clinical Trial Number | Phase | Purpose | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NCT04427072 | Phase III | Previously treated advanced NSCLC patients with MET exon 14 skipping mutation treated with capmatinib versus docetaxel | |||||||
| NCT04926831 | Phase II | Efficacy and safety of neoadjuvant and adjuvant capmatinib | |||||||
| Noncardiac chest pain | |||||||||
| NCT05435846 | 0 | 8 (12) | 1 (7) | 4 (10) | 8 (15) | 8 (27) | |||
| Overall response | |||||||||
| No. of patients with overall response | |||||||||
| Decreased appetite | 76 (21) | 3 (1) | |||||||
| No. of patients with disease control | 54 | 27 | 49 | ||||||
| Constipation | 66 (18) | 10 | 23 | 25 | 16 | ||||
| 3 (1) | Percent of patients (95% CI) | 78 (67–87) | 96 (82–100) | 71 (59–81) | 67 (38–88) | 55 (39–70) | 46 (33–60) | ||
| Diarrhea | 53 (34–72) | ||||||||
| 64 (18) | 2 (1) | Duration of Response | |||||||
| Cough | 58 (16) | No. of events/No. of patients with response | 23/28 | 11/19 | 15/20 | 6/6 | 3/5 | 4/5 | 2/2 |
| Median duration of response (95% CI)—mo | 9.7 (5.6–13.0) | 12.6 (5.6–NE) | 8.3 (4.2–15.4) | 7.5 (2.6–14.3) | 24.9 (2.7–24.9) | 9.7 (4.2–NE) | 4.2 (4.2–4.2) | ||
| Progression-free survival | |||||||||
| 35 (10) | 4 (1) | ||||||||
| Phase I/Ib | Serious adverse event—No. (%) | 184 (51) | 152 (42) | ||||||
| Capmatinib plus trametinib in patients with | MET | exon 14 skipping mutation | |||||||
| NCT04677595 | Phase II | Chinese patients who are EGFR wt and ALK rearrangement negative with MET exon 14 skipping mutation | |||||||
| 58 | 15 | 34 | 50 | 22 | |||||
| Median progression-free survival (95% CI)—mo | 5.4 (4.2–7.0) | 12.4 (8.2–NE) | |||||||
| Event leading to discontinuation—No. (%) | 56 (15) | 35 (10) |
3
| NCT05110196 | ||
| Phase IV | ||
| Indian patients with | MET | exon 14 skipping mutation |
| NCT05488314 | Phase I/II | Combination therapy of capmatinib and amivantamab in unresectable Stage IV NSCLC in patients with MET exon 14 skipping mutations or MET amplification |
| NCT05642572 | Phase II | Combination therapy of capmatinib with osimertinib +/− ramucirumab in EGFR mutant, MET-amplified, Stage IV or recurrent NSCLC |
| 4.1 (2.9–4.8) | ||
| 4.2 (1.4–6.9) | ||
| 2.7 (1.4–3.1) | ||
| 2.7 (1.4–4.1) | ||
| 3.6 (2.2–4.2) |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316.
- Shaw, A.T.; Riely, G.J.; Bang, Y.-J.; Kim, D.-W.; Camidge, D.R.; Solomon, B.J.; Varella-Garcia, M.; Iafrate, A.J.; Shapiro, G.I.; Usari, T.; et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): Updated results, including overall survival, from PROFILE 1001. Ann Oncol. 2019, 30, 1121–1126.
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.-J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 261–270.
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824.
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14–Mutated or MET -Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957.
- Ramalingam, S.S.; Yang, J.C.-H.; Lee, C.K.; Kurata, T.; Kim, D.-W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849.
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381.
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakeley, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282.
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2 -Mutant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251.
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45.
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signalling pathway in cancer. Eur. J. Cancer 2010, 46, 1260–1270.
- Pilotto, S.; Carbognin, L.; Karachaliou, N.; Ma, P.C.; Rosell, R.; Tortora, G.; Bria, E. Tracking MET de-addiction in lung cancer: A road towards the oncogenic target. Cancer Treat. Rev. 2017, 60, 1–11.
- Lee, M.; Jain, P.; Wang, F.; Ma, P.C.; Borczuk, A.; Halmos, B. MET alterations and their impact on the future of non-small cell lung cancer (NSCLC) targeted therapies. Expert Opin. Ther. Targets 2021, 25, 249–268.
- Salgia, R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol. Cancer Ther. 2017, 16, 555–565.
- Tu, C.-Y.; Cheng, F.-J.; Chen, C.-M.; Wang, S.-L.; Hsiao, Y.-C.; Chen, C.-H.; Tsia, T.-C.; He, Y.-H.; Wang, B.-W.; Hsieh, I.-S.; et al. Cigarette smoke enhances oncogene addiction to c-MET and desensitizes EGFR-expressing non-small cell lung cancer to EGFR TKIs. Mol. Oncol. 2018, 12, 705–723.
- Cheng, F.-J.; Chen, C.-H.; Tsai, W.-C.; Wang, B.-W.; Yu, M.-C.; Hsia, T.-C.; Wei, Y.-L.; Hsiao, Y.-C.; Hu, D.-W.; Ho, C.-Y.; et al. Cigarette smoke-induced LKB1/AMPK pathway deficiency reduces EGFR TKI sensitivity in NSCLC. Oncogene 2021, 40, 1162–1175.
- Hong, L.; Zhang, J.; Heymach, J.V.; Le, X. Current and future treatment options for MET exon 14 skipping alterations in non-small cell lung cancer. Ther. Adv. Med. Oncol. 2021, 13, 1758835921992976.
- Cortot, A.B.; Kherrouche, Z.; Descarpentries, C.; Wislez, M.; Baldacci, S.; Furlan, A.; Tulasne, D. Exon 14 Deleted MET Receptor as a New Biomarker and Target in Cancers. JNCI J. Natl. Cancer Inst. 2017, 109, djw262.
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non–Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5, 653–663.
- Tong, J.H.; Yeung, S.F.; Chan, A.W.H.; Chung, L.Y.; Chau, S.L.; Lung, R.W.M.; Tong, C.Y.; Chow, C.; Tin, E.K.Y.; Yu, Y.H.; et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non-Small Cell Lung Carcinoma with Poor Prognosis. Clin. Cancer Res. 2016, 22, 3048–3056.
- Schubart, C.; Stöhr, R.; Tögel, L.; Fuchs, F.; Sirbu, H.; Seitz, G.; Seggewiss-Bernhardt, R.; Leistner, R.; Sterlacci, W.; Vieth, M.; et al. MET Amplification in Non-Small Cell Lung Cancer (NSCLC)—A Consecutive Evaluation Using Next-Generation Sequencing (NGS) in a Real-World Setting. Cancers 2021, 13, 5023.
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737.
- Chmielecki, J.; Mok, T.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.-H.; Shepherd, F.A.; et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell lung cancer from the AURA3 trial. Nat Commun. 2023, 14, 1071.
- Dagogo-Jack, I.; Yoda, S.; Lennerz, J.K.; Langenbucher, A.; Lin, J.J.; Rooney, M.M.; Prutisto-Chang, K.; Oh, A.; Adams, N.A.; Yeap, B.Y.; et al. MET Alterations Are a Recurring and Actionable Resistance Mechanism in ALK-Positive Lung Cancer. Clin. Cancer Res. 2020, 26, 2535–2545.
- Awad, M.M.; Leonardi, G.C.; Kravets, S.; Dahlberg, S.E.; Drilon, A.; Noonan, S.A.; Camidge, D.R.; Ou, S. H-I; Costa, D.B.; Gadgeel, S.M.; et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: A retrospective analysis. Lung Cancer 2019, 133, 96–102.
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Janne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730.
- Bahcall, M.; Paweletz, C.P.; Kuang, Y.; Taus, L.J.; Sim, T.; Kim, N.D.; Dholakia, K.H.; Lau, C.J.; Gokhale, P.C.; Chopade, P.R.; et al. Combination of Type I and Type II MET Tyrosine Kinase Inhibitors as Therapeutic Approach to Prevent Resistance. Mol. Cancer Ther. 2022, 21, 322–335.
- Cui, J.J. Targeting receptor tyrosine kinase MET in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453.
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, N.; et al. Once-daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non-small-cell lung cancers harbouring MET exon 14 skipping alterations: A multicentre, single-arm, open-label, phase 2 study. Lancet Respir. Med. 2021, 9, 1154–1164.
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943.
- Santarpia, M.; Massafra, M.; Gebbia, V.; D’Aquino, A.; Garipoli, C.; Altavilla, G.; Rosell, R. A narrative review of MET inhibitors in non-small cell lung cancer with MET exon 14 skipping mutations. Transl. Lung Cancer Res. 2021, 10, 1536–1556.
- Yan, S.B.; Um, S.L.; Peek, V.L.; Stephens, J.R.; Zeng, W.; Konicek, B.W.; Ling, L.; Manro, J.R.; Wacheck, W.; Walgren, R.A. MET-targeting antibody (emibetuzumab) and kinase inhibitor (merestinib) as single agent or in combination in a cancer model bearing MET exon 14 skipping. Investig. New Drugs 2018, 36, 536–544.
- Calles, A.; Kwiatkowski, N.; Cammarata, B.K.; Ercan, D.; Gray, N.S.; Jänne, P.A. Tivantinib (ARQ 197) efficacy is independent of MET inhibition in non-small-cell lung cancer cell lines. Mol. Oncol. 2015, 9, 260–269.
- Klempner, S.J.; Borghei, A.; Hakimian, B.; Ali, S.M.; Ou, S.-H.I. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. J. Thorac. Oncol. 2017, 12, 152–156.
- Scagliotti, G.; von Pawel, J.; Novello, S.; Ramlau, R.; Favaretto, A.; Barlesi, F.; Akerley, W.; Orlov, S.; Santoro, A.; Spigel, D.; et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2667–2674.
- Baltschukat, S.; Engstler, B.S.; Huang, A.; Hao, H.-X.; Tam, A.; Wang, H.Q.; Liang, J.; DiMare, M.T.; Bhang, H.-E.C.; Wang, Y.; et al. Capmatinib (INC280) Is Active Against Models of Non–Small Cell Lung Cancer and Other Cancer Types with Defined Mechanisms of MET Activation. Clin. Cancer Res. 2019, 25, 3164–3175.
- Novartis. TABRECTA (Capmatinib): US Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213591s000lbl.pdf (accessed on 8 March 2023).
- Dhillon, S. Capmatinib: First Approval. Drugs 2020, 80, 1125–1131.
More