Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Fabio Polticelli.
Iron is an essential transition metal for its involvement in several crucial biological functions, the most notable being oxygen storage and transport. Due to its high reactivity and potential toxicity, intracellular and extracellular iron levels must be tightly regulated. This is achieved through transport systems that mediate cellular uptake and efflux both at the level of the plasma membrane and on the membranes of lysosomes, endosomes and mitochondria.
- iron transport
- ferroportin
- transferrin
- DMT1
1. General Iron Metabolism and Trafficking
Among all metals, iron is probably the one that most exhibits a double face in living organisms: essential for numerous biological functions, from oxygen and electron shuttling to DNA synthesis and repair [1]; harmful because of its ability to monoelectronically reduce oxygen, triggering physiologically defensive or pathologically dangerous oxidative stress cascades [2,3][2][3].
In addition to, and in conjunction with, possessing this two-faced behavior, iron is also difficult for cells to handle. In fact, the largely prevalent form in the aerobic environment is the ferric ion Fe3+. However, Fe3+ salts are very poorly soluble in polar solvents like water. All organisms, therefore, have evolved mechanisms of iron import, export and storage aimed, on the one hand, at accumulating and utilizing the metal in bioavailable forms and, on the other hand, at keeping its oxidoreductive exuberance under control.
Mammals have no regulated pathways of iron excretion, so the amount of metal absorbed with the diet must balance the iron lost by bleeding or by sloughing of epithelial cells. An adult human body contains 3–4 g of iron, and daily losses amount to 1–2 mg. In mammalian cells, iron is mainly distributed in mitochondria (ca. 16 μM), cytosol (ca. 6 μM), nuclei (ca. 7 μM) and lysosomes (ca 16 μM) [4]. Therefore, all cells contain and use iron. However, overall metal homeostasis is governed primarily by very few cellular types: erythrocytes and their precursors, which contain about two-thirds of all the iron in the body; hepatocytes and macrophages, which are reservoir sites for the metal; and enterocytes, which, along with macrophages, provide the gateway for the metal to enter the circulatory system, the blood being the last of the compartments with a key role in the iron game.
Uptake of dietary inorganic iron is due to the presence of DMT1 (divalent metal transporter 1) on the apical side of enterocytes [5]. This transporter handles reduced Fe2+ as a substrate, but most dietary iron is in the oxidized Fe3+ form. Therefore, iron has to be reduced before it can be absorbed. This is achieved through duodenal cytochrome B (CYBRD1) and possibly other reductases [6].
For iron to cross the basolateral membrane of enterocytes and enter the bloodstream, a dual system is in place, constituted by ferroportin (FPN), the sole known exporter of iron in multicellular organisms, and hephaestin (HEPH), a multicopper ferroxidase required to load the metal onto transferrin, the iron carrier of the blood [7]. Most of the circulating iron, however, does not come from dietary absorption, but rather from metal recycling, with splenic macrophages playing a major role as collectors of iron-rich senescent erythrocytes. Macrophages also use FPN to load Fe3+ onto circulating transferrin; in this case, the ferroxidase partner is ceruloplasmin (CP), a paralog of HEPH [8]. It is important to mention that FPN has been recently shown to be highly abundant on red blood cell membrane [9]. Multiple pieces of evidence point to the role of erythroid cells FPN in maintaining systemic iron homeostasis and in protecting red blood cells from oxidative stress [9,10][9][10].
Transferrin (TF) is an 80-kDa bilobal glycoprotein synthesized by hepatocytes and consisting of two homologous N- and C-lobes. Each lobe contains a high-affinity (Kd ≅ 10−20 M) [11] iron-binding site formed by four amino acid ligands: a histidine, an aspartate and two tyrosine residues. Metal binding also requires the presence of a carbonate anion, anchored to a conserved arginine residue, that occupies the two remaining coordination sites. Selective iron delivery to cells in peripheral tissues occurs through the specific interaction of Fe-TF with its receptor TFRC, a type II transmembrane homodimeric glycoprotein. The TF-TFRC complex readily enters cells by clathrin-mediated endocytosis. The interaction between TFRC and TF is dependent on pH; at pH 7.4, TFRC binds to iron-loaded holo-TF but not to iron-free apo-TF. In contrast, at a lower pH in the endosome, TFRC binds to apo-TF but not to holo-TF [12]. In the endosome, acidification allows the release of Fe3+, which is reduced to Fe2+ by the ferrireductase STEAP3 [13] and poured into the cytoplasm by DMT1 [14], and probably by ZIP14 [15] and by type IV mucolipidosis-associated protein TRPML1 [16]. Iron-free TF remains bound to TFRC in the acidified endosome and is finally recycled, returning to the cell surface where the complex dissociates. Corollary to this mechanism is the fact that only a fraction of transferrin circulates in the blood bound to iron: in fact, the average saturation of the protein is around 30 percent.
A second receptor for TF has been identified about two decades ago (see reference [17] for a review). TFR2 shares structural and functional similarities with TFRC, despite that its regulatory machine appears peculiar. Hereditary hemochromatosis protein (HFE), a protein belonging to the family of major histocompatibility complex class I molecules, competes with TF for TFR2 and may thereby block the binding of Fe-TF to TFR2, preventing internalization of the complex and negatively regulating cellular uptake of TF-bound iron.
Several cell types, including neurons, hepatocytes and cardiomyocytes, can uptake iron in the form of the so-called non-transferrin-bound iron (NTBI), a potentially toxic form of the metal that results from pathological excessive entry of iron into plasma, for example in hereditary hemochromatosis and thalassemia major. Under these conditions, the saturation capacity of transferrin is exceeded and iron complexes with a number of low-molecular weight molecules, as well nonspecifically with other plasma proteins. Plasma NTBI is rapidly cleared by most cells through less selective but highly efficient iron transporters, including SLC39A14 (ZIP14 [18]), SLC39A8 (ZIP8 [19]), DMT1 [20], L-type Ca2+ channels (LTTCs [21]) and T-type Ca2+ channels (TTCCs [22,23][22][23]). Comprehensive reviews on NTBI transporters can be found in the literature [24,25][24][25]. A large part of the ferrous NTBI is translocated into the mitochondria for the biogenesis of heme and Fe-S clusters. This is mainly allowed by mitoferrin (MFRN), a small transmembrane transporter (~300 residues) located on the inner mitochondrial membrane [26]. A role in mitochondrial iron trafficking has also been shown for ZIP8 [27] and DMT1 [28,29,30][28][29][30]. A general scheme of cellular iron traffic is depicted in Figure 1.
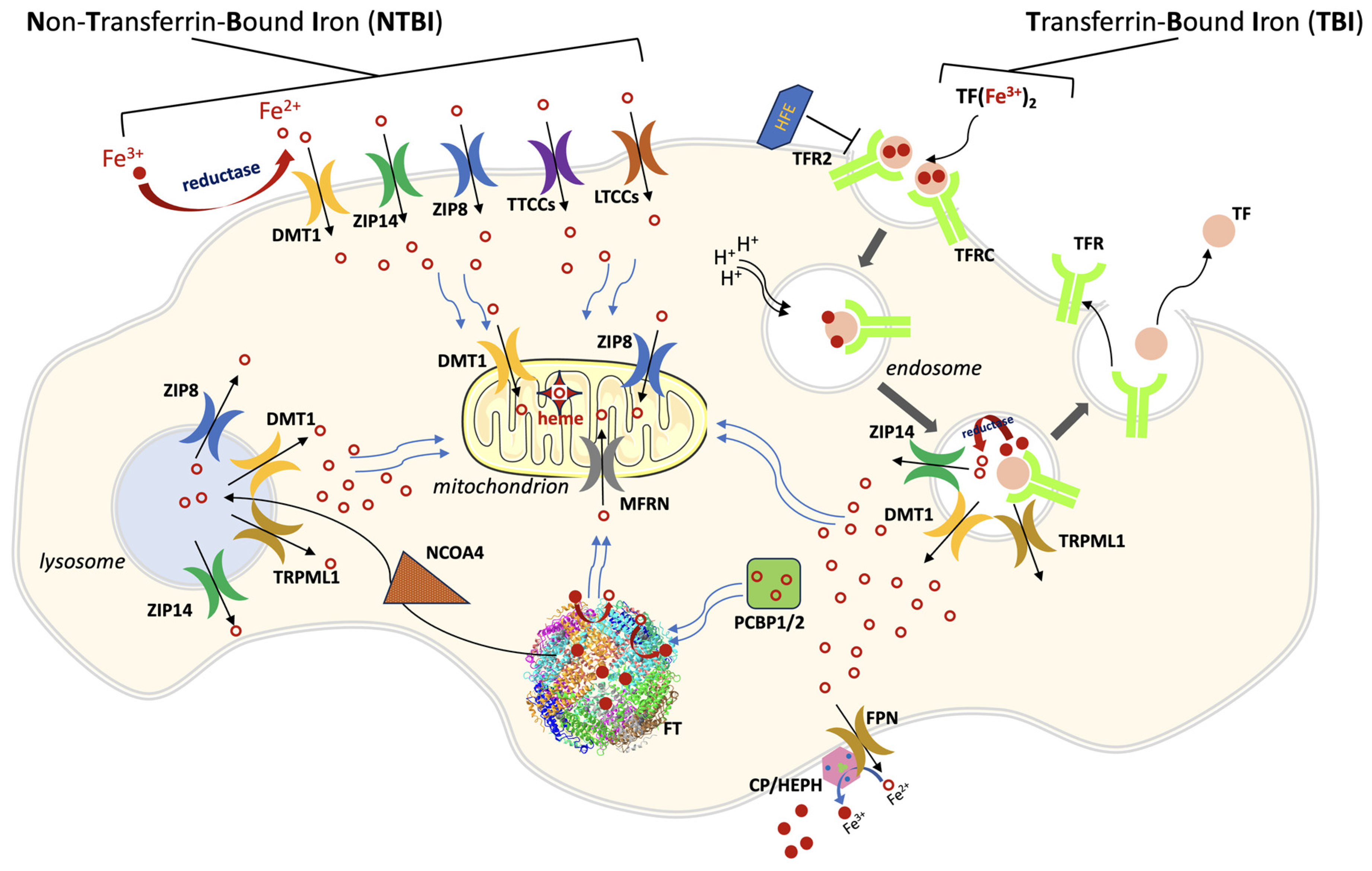
Figure 1. A general scheme of cellular iron traffic. Iron can enter cells both as TBI (transferrin-bound iron) or as NTBI (non-transferrin-bound iron). Iron-loaded transferrin binds to its ubiquitous receptor TFRC (and in some cell types to TFR2) on the cell membrane and the complex is endocytosed. The endosome is acidified, iron is released from TF and poured into cytoplasm through divalent metal transporter 1 (DMT1), and possibly through ZIP14 and type IV mucolipidosis-associated protein TRPML1. The empty complex dissociates and is then recycled outside the cell for a new transport cycle. NTBI circulates in the plasma as Fe3+ in low-molecular weight complexes or bound to albumin. After a reduction step, it is taken up as Fe2+ through different importers, depending on the cell type: ZIP14, ZIP8, DMT1, LTCCs (L-type Ca2+ channels), TTCCs (T-type Ca2+ channels). ZIP8 and ZIP14 are also present on lysosomal, mitochondrial, and endosomal membranes. Iron trafficking in the cytosol is mediated by PCBP1/2 (poly(rC) binding protein 1/2), which cargos the metal to ferritin (FT), the iron storage protein, and to other iron-containing enzymes (not shown). In the mitochondrion, the main iron-transport systems are constituted by mitoferrin 1/2 (MFRN) on the inner membrane, and by DMT1 and ZIP8 on the outer membrane. Ferritin iron stores are mobilized through the action of Nuclear Receptor Coactivator 4 (NCOA4), which directs ferritin to the lysosome for degradation. Lysosomes pour iron into cytoplasm through at least four exporters (ZIP8, ZIP14, DMT1 and TRPML1). Cellular iron export is mediated by ferroportin (FPN), which acts in concert with a multicopper ferroxidase (CP, ceruloplasmin or HEPH hephaestin, depending on the cell type).
When intracellular iron levels exceed metabolic needs, two mechanisms intervene to prevent the unwanted presence of the metal and its potentially toxic consequences. On the one hand, intracellular levels of ferritin (FT), a multimeric protein composed of 24 subunits that functions as a veritable cage for iron, increase. Over 4000 iron atoms can be sequestered in a redox-inactive form by FT inside its core [31], with the iron chaperones PCBP1 and PCBP2 enhancing the process [32,33][32][33]. On the other hand, levels of FPN on the plasma membrane also increase. As expected, when the cell goes into iron deficiency, the opposite mechanisms are triggered: levels of the TF receptor TFRC increase and synthesis of FT and FPN decreases. This is mainly mediated at the translational level by the presence on the corresponding mRNAs of iron-responsive elements (IREs) which govern the rate of protein synthesis [34]. Very recently, a connection between the iron saturation rate of TF and the levels of FPN on the cell membrane in endothelial cells has been reported [35].
Systemic iron levels must also be tightly regulated. Iron overload leads to hemochromatosis, a detrimental condition that affects parenchymal organs including the liver, heart, and pancreas [36]. Iron deficiency leads to anemia, a global health problem with several pathological consequences including cognitive developmental defects in children, poor physical performance, and unfavorable pregnancy outcomes [37]. It has long been known that, under physiological conditions, blood iron levels are remarkably stable, even when dietary intake is highly variable. It is also known that intestinal absorption of iron increases several times under conditions of metal deficiency and decreases when iron is in excess, demonstrating that iron homeostasis is under endocrine control. The key player is in fact hepcidin (HAMP), a small 25-aa peptide hormone synthesized in the liver [38], that acts as an inhibitor of FPN and whose levels are positively or negatively regulated at the transcriptional level depending on physiological conditions. Activation of the immune system leads to iron retention in macrophages and reduced dietary iron absorption as the result of a massive production of several pro-inflammatory cytokines. Among these, interleukin 6 (IL6) is the main stimulator of the hepatic synthesis of hepcidin through STAT3 [39]. Levels of TF saturation and of intracellular iron stores also positively regulate the synthesis of hepcidin. On the other hand, activation of erythropoiesis leads to the synthesis of the erythroid factor erythroferrone (ERFE), which is a negative regulator of hepcidin [39]. As detailed below, the hormone silences FPN by physically interacting with it and decreases its activity by two mechanisms: on the one hand, it binds in the central cavity of the transporter inhibiting the passage of iron [40]; on the other, binding of hepcidin in a specific pocket of FPN induces its internalization and degradation [41].
2. Ferroportin
Ferroportin (FPN) is the only known human iron exporter [42]. Mutations of FPN cause type 4 haemochromatosis, a disease characterized by two different iron accumulation phenotypes [43]. These different phenotypes are the result of loss-of-function mutations, which impair the transport activity, or gain-of-function mutations, affecting hepcidin-mediated FPN internalization and degradation [41]. The first attempts to shed light on FPN structure through molecular modelling techniques revealed that FPN is a member of the Major Facilitator Superfamily (MFS) [44,45,46][44][45][46]. MFS members represent the largest class of secondary active transporters [47,48][47][48]. They share a conserved fold, characterized by 12 transmembrane helices forming an N-terminal and a C-terminal domain, each organized into a pair of inverted 3-plus-3 helices repeats, connected by a cytoplasmic loop [48]. In the N-domain, helices 1, 2 and 3 are related to helices 4, 5 and 6 by a 180° rotation around an axis parallel to the membrane bilayer. Similarly, in the C-domain, helices 7, 8, 9 are related to helices 10, 11 and 12 [49] (Figure 2).
The FPN transport cycle occurs through a process known as alternating access mechanism, in which conformational changes between different states (inward-open, occluded, and outward-open) take place. This process relies on the rigid-body relative rotation of the N-terminal and C-terminal domains of FPN, as depicted in Figure 3 [48,49,50][48][49][50]. During this process, the first set of helices (helices 1, 4, 7 and 10) and second set of helices (helices 2, 5, 8 and 11) within each repeat of the protein, which in the inward-open state interact along the cytoplasmic ends, undergo a conformational change and switch their interaction from the cytoplasmic to the extracellular ends of the protein. This alternating access mechanism, driven by the rotation of the N-terminal and C-terminal domains, enables the movement of substrates (such as iron) across the cell membrane.
It has been demonstrated that FPN acts through an electroneutral export mechanism by coupling the export of one Fe2+ ion to the import of two H+ [51], although other data suggest that FPN may be a Fe2+/H+ symporter [52]. Structural studies of mammalian FPN evidenced the presence of two metal binding sites [51[51][53],53], the first composed by Asp39 and His43, and the second by Cys326 and His507. Recently, structural and functional studies demonstrated that FPN can mediate Ca2+ influx through a uniport mechanism and that Asp39 is also involved in binding this metal. In fact, Ca2+ is observed to be bound in the vicinity of Asp39 and the Asp39Ala mutation abolishes Ca2+ transport [54]. While Fe2+ ions inhibit Ca2+ transport, the opposite does not hold. However, the physiological significance of Ca2+ transport by FPN is still unclear.
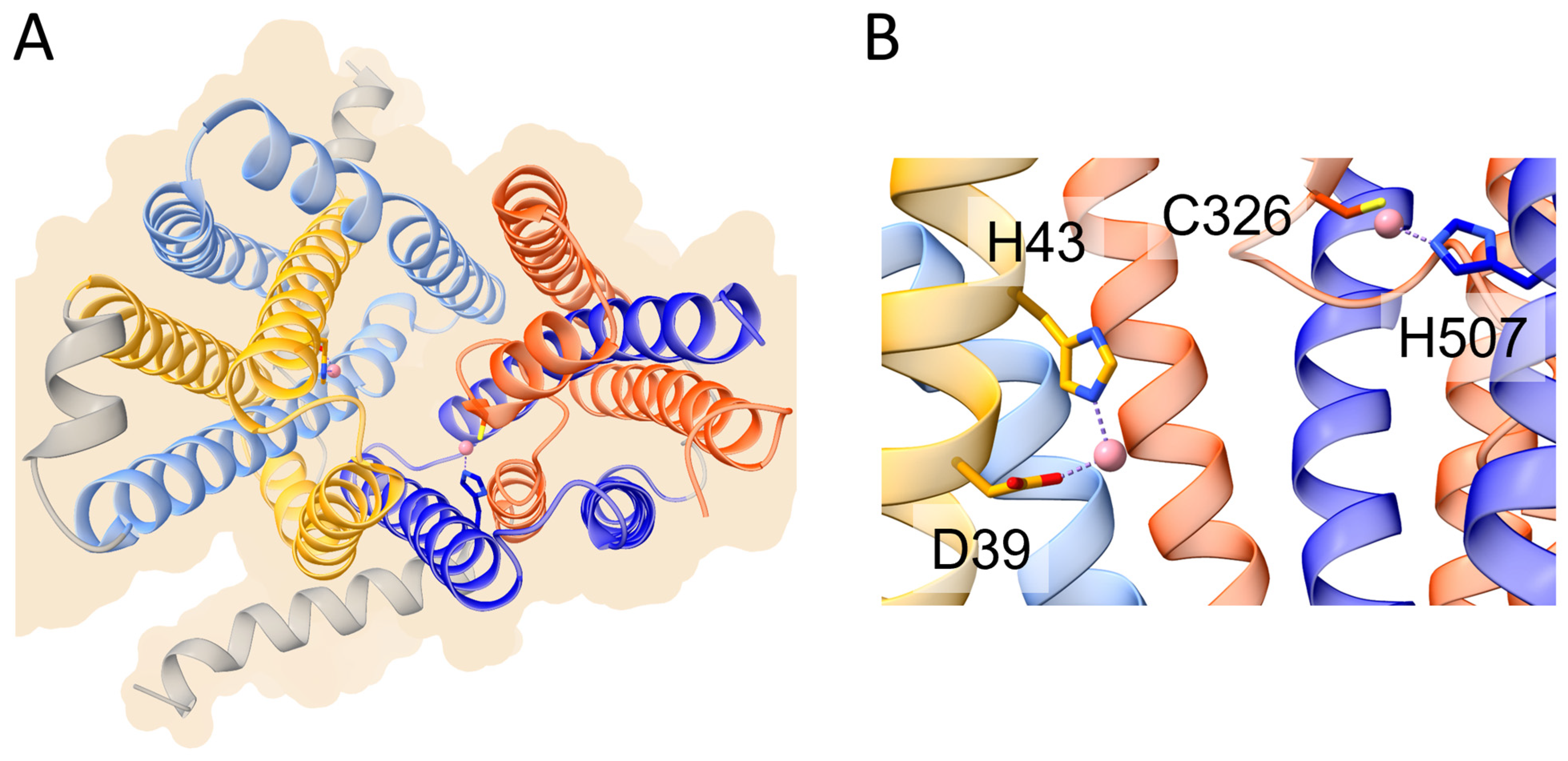
Figure 2. Schematic representation of the overall structure and metal binding sites of human ferroportin (PDB ID 8Dl8). (A) Overall structure of human ferroportin shown in ribbon representation. The helices of the first and second inverted repeats of the N-domain are colored in yellow and cornflower blue, respectively. The helices of the corresponding inverted repeats of the C-domain are colored in orange and blue, respectively (see text for details). (B) Metal binding sites on the N-domain (left) and C-domain (right). The cobalt ions mimicking iron are shown as pink spheres. Metal ligands are shown in stick representation.
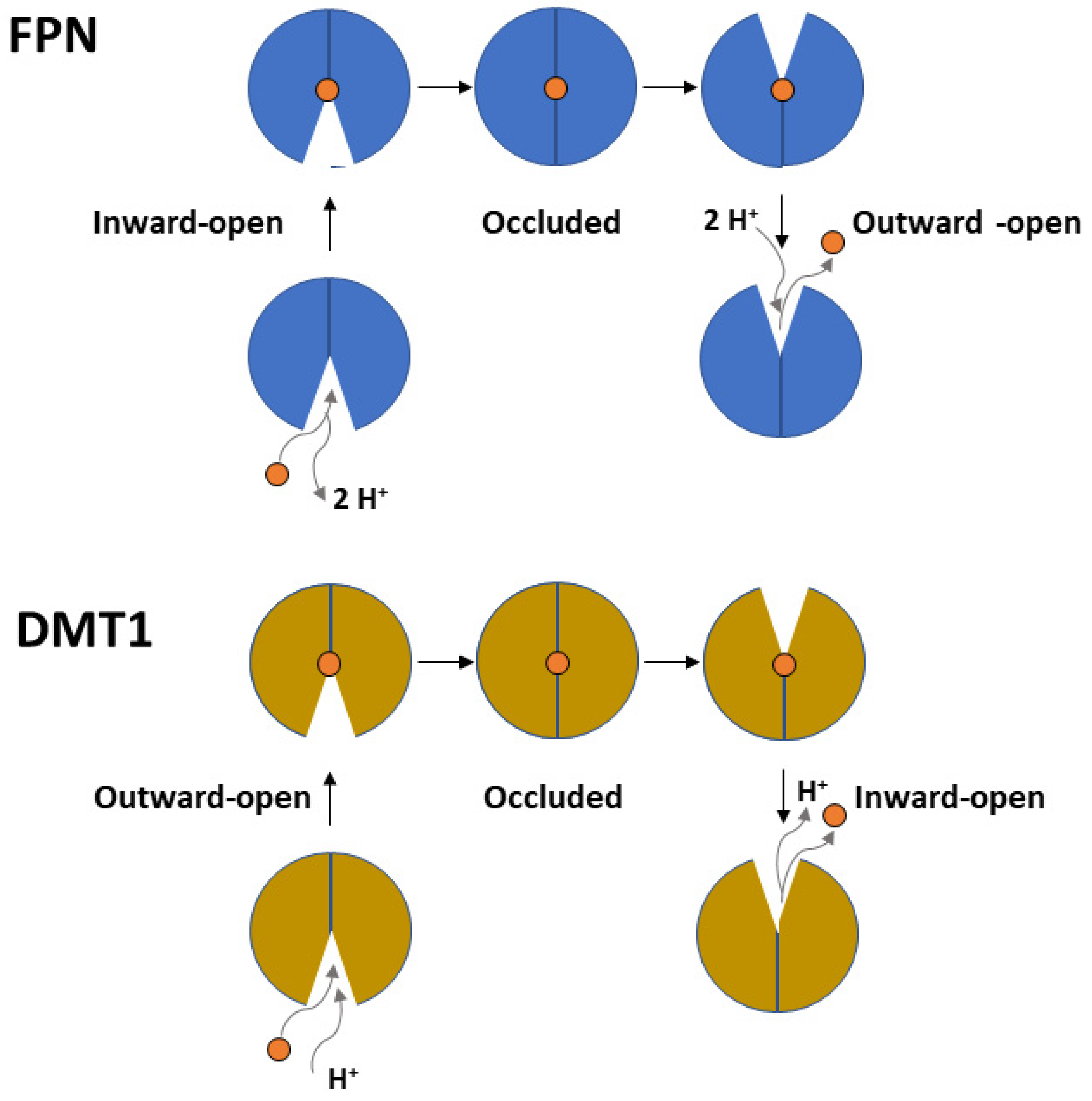
Figure 3. Top panel. Scheme depicting the proton-coupled, electroneutral Fe2+ transport mediated by FPN through cycling from an inward-open to an occluded and outward-open conformation. Bottom panel. Similar scheme depicting the proton-coupled-symport of Fe2+ mediated by DMT1.
Hemochromatosis type 4 is an autosomal dominantly inherited iron overload pathology caused by the mutation of FPN. Two distinct phenotypes are observed, where iron overload affects different organs: mainly reticulo-endothelial macrophages with low transferrin saturation (type 4A) or liver hepatocytes with high transferrin saturation (type 4B) [55,56][55][56]. Over 60 missense mutations have been identified in FPN and they have been classified as either loss-of-function or gain-of-function. Loss-of-function mutations cause hemochromatosis type 4A, also named ‘ferroportin disease’, while gain-of-function variants give rise to hemochromatosis type 4B. Loss of function is due to either a direct impact on the capacity of the protein to transport iron or to a folding/subcellular localization defect. Impairment of iron transport can derive from decreased iron binding, such as for the Asp181Val/Asn mutant [57], and/or from the alteration of interactions necessary for the conformational switch required for the translocation of iron across the membrane. An example is the Arg178Gln mutation that disrupts an inter-lobe salt bridge with Asp473, causing destabilization of the outward open state of FPN [58]. Of note, all residues composing motif A (Gly80-Asp84-Arg88) and Asp157, which establish a network of interactions that stabilize the outward-open conformation of FPN, have been found to be mutated in patients, clearly indicating a critical role for conformational transitions in the iron transport cycle [56,59][56][59]. Variants Asp157Gly/Tyr/Asn are classified as loss-of-function, while the newly discovered Asp157Ala one appears to lead to a gain-of-function [60], highlighting the difficulty in predicting the effect of mutations. Gain-of-function mutations lead to resistance of FPN to hepcidin, either by the inability to bind the peptide in the central cavity of FPN in the outward conformation or by altering the internalization/ubiquitination process. Interestingly, Cys326Ser/Tyr/Phe and His507Arg mutations, which target residues directly involved in iron binding, are classified as gain-of-function because they also lead to hepcidin resistance, possibly due to the overlap of binding sites for the metal and the peptide. The experimentally determined three-dimensional structures of human FPN have allowed to most mutations to be mapped and a clearer picture of their molecular impact to be obtained. However, full understanding is still lacking given the fact that some mutations are found in regions of the protein that are not resolved, and that structures of FPN are available for the outward-open and occluded states but not for the inward-open conformation. A quite complete, though non-exhaustive list of gain-of-function and loss-of-function mutations with a discussion on their pathogenic role can be found in Tortosa et al., 2017 and references therein [59].
Among gain-of-function mutations, those affecting internalization/ubiquitination of FPN are the most intriguing and are less understood from a molecular point of view. In fact, the details of the process of ubiquitination and internalization have been only partially deciphered. Lysine residue(s) located in the unresolved long intracellular loop connecting the N- and C-terminal domains of FPN are the target for polyubiquitin addition. Evidence suggesting the E3 ubiquitin-ligase Rnf217 [61] and the E1 enzyme UBA6 together with the adaptor protein NDFIP1 [62] are involved in degradation of FPN has been recently reported. The redundancy of pathway components makes it challenging to exactly define which target lysines on FPN and which ubiquitinating enzymes are required for hepcidin-dependent degradation of the transporter. To further complicate matters, FPN has also been reported to be subjected to sumoylation, with the gain-of-function mutation Lys240Glu disrupting this process [63].
Another physiologically relevant aspect that is beginning to be elucidated is the identity of the intracellular donor of ferrous iron to FPN. Recently, it has been determined that PCBP2, originally described as poly(rC) RNA binding protein 2, is in fact a dual-function protein, acting also as an iron chaperone [32]. PCBP2 is a ubiquitously expressed cytosolic protein composed of three KH domains. FPN was demonstrated to interact with the iron-loaded form of PCBP2 only. The iron-dependent interaction was determined to involve the cytoplasmic C-terminal region of FPN and the KH2 domain of PCBP2 [64]. Remarkably, PCBP2 directly links iron uptake to iron export in the cell: the iron chaperone receives the metal from DMT1 (see below) and delivers it to the iron exporter FPN.
3. DMT1
DMT1 belongs to the ubiquitous solute carrier 11 (SLC11) family of transporters. In vertebrates, DMT1 (SLC11A2) is the primary transporter of Fe2+ from the intestinal lumen to the cytoplasm of enterocytes. In addition, DMT1 is also responsible for the transport of Fe2+ from the interior of intracellular vesicles to cytoplasm (see Manatschal and Dutzler, 2022 [65] for a recent comprehensive review covering the structural and functional aspects). In both cases, iron transport is coupled to co-transport of H+ [66]. Interestingly, DMT1 localization on the outer membrane of mitochondria has also been evidenced [28[28][29],29], and a recent study demonstrated that DMT1 is involved in mitochondrial uptake of Fe2+ and Mn2+ [30]. Indeed, several studies evidenced the rather broad specificity of human DMT1 that can also transport Co2+, Mn2+, Cd2+ and Ni2+, although Fe2+ is the preferred substrate [67]. From a structural viewpoint, studies on the bacterial orthologs of the DMT1/NRAMP family identified in Staphylococcus capitis [68], Deinococcus radiodurans [69,70][69][70] and Escherichia coli [71] revealed a fold first observed in the Aquifex aeolicus LeuT amino acid transporter. The main characteristic of this fold is the presence of twelve transmembrane helices (TM), of which the first ten are organized into two domains (TM1–5 and TM6–10) related by a pseudo-two-fold axis in the membrane plane [72] (Figure 4). Interestingly, the central substrate binding site is made up by residues located on TM1 and TM6 in unwound regions [73]. As already described for the MFS members like FPN, the transport cycle involves conformational changes between an outward-open and an inward-open state, through an intermediate occluded state (Figure 3) [65]. Very recently, novel evidence on the metal transport pathway of the members of the DMT1/NRAMP has been obtained through structural analysis of Deinococcus radiodurans NRAMP bound to Mn2+ in all the three relevant conformational states [73]. The structures clearly highlight how global conformational changes are driven by changes in the coordination sphere of the metal, made possible by the relatively high flexibility of the TM1 and TM6 helices on which metal binding residues are located (Figure 4).
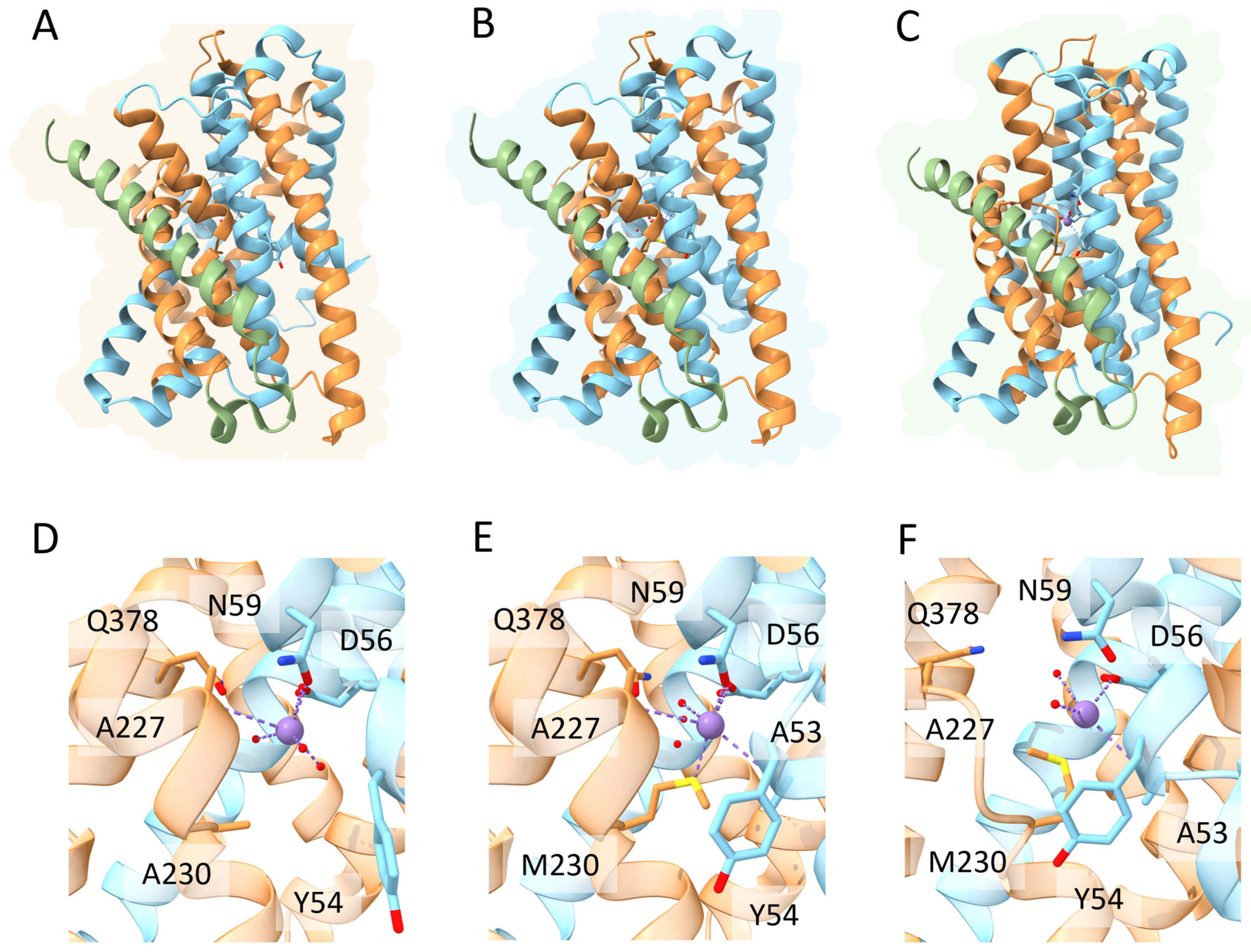
Figure 4. Schematic representation of the relevant conformational states and metal binding sites of Deinococcus radiodurans NRAMP. Top panels. Overall structure of the (A) inward-open; (B) occluded and (C) outward open conformational states of the transporter (PDB IDs 8E6I, 8E60 and 8E6N, respectively). The first five helices are colored in gold, the second five helices in cyan and the additional C-terminal helix in green (see text for details). Bottom panels. Detailed view of the Mn2+ ion coordination sphere in the (D) inward-open; (E) occluded and (F) outward open conformational states. Mn2+ ion is shown as a purple sphere, water molecules as small red spheres and coordinating residues in stick representation.
References
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38.
- Anderson, G.J.; Frazer, D.M. Current Understanding of Iron Homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S.
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria Regulation in Ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058.
- Huo, C.; Li, G.; Hu, Y.; Sun, H. The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation. Int. J. Mol. Sci. 2022, 23, 14195.
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; DiRenzo, C.; Robine, S.; Andrews, N.C. Slc11a2 Is Required for Intestinal Iron Absorption and Erythropoiesis but Dispensable in Placenta and Liver. J. Clin. Invest. 2005, 115, 1258–1266.
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759.
- Fuqua, B.K.; Lu, Y.; Darshan, D.; Frazer, D.M.; Wilkins, S.J.; Wolkow, N.; Bell, A.G.; Hsu, J.; Yu, C.C.; Chen, H.; et al. The Multicopper Ferroxidase Hephaestin Enhances Intestinal Iron Absorption in Mice. PLoS ONE 2014, 9, e98792.
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-Ferroportin System of Iron Traffic in Vertebrates. World J. Biol. Chem. 2014, 5, 204–215.
- Zhang, D.-L.; Wu, J.; Shah, B.N.; Greutélaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic Ferroportin Reduces Intracellular Iron Accumulation, Hemolysis, and Malaria Risk. Science 2018, 359, 1520–1523.
- Zhang, D.-L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin Deficiency in Erythroid Cells Causes Serum Iron Deficiency and Promotes Hemolysis Due to Oxidative Stress. Blood 2018, 132, 2078–2087.
- Aisen, P.; Leibman, A.; Zweier, J. Stoichiometric and Site Characteristics of the Binding of Iron to Human Transferrin. J. Biol. Chem. 1978, 253, 1930–1937.
- Enns, C.A.; Rutledge, E.A.; Williams, A.M. The Transferrin Receptor. In Biomembranes: A Multi-Volume Treatise; Lee, A.G., Ed.; Endoctosis and Exocytosis: Amsterdam, The Netherlands, 1996; Volume 4, pp. 255–287.
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a Ferrireductase Required for Efficient Transferrin-Dependent Iron Uptake in Erythroid Cells. Nat. Genet. 2005, 37, 1264–1269.
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 Is Mutated in the Anemic Belgrade (b) Rat: Evidence of a Role for Nramp2 in Endosomal Iron Transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153.
- Zhao, N.; Gao, J.; Enns, C.A.; Knutson, M.D. ZRT/IRT-like Protein 14 (ZIP14) Promotes the Cellular Assimilation of Iron from Transferrin*. J. Biol. Chem. 2010, 285, 32141–32150.
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The Type IV Mucolipidosis-Associated Protein TRPML1 Is an Endolysosomal Iron Release Channel. Nature 2008, 455, 992–996.
- Kawabata, H. Transferrin and Transferrin Receptors Update. Free Radic. Biol. Med. 2019, 133, 46–54.
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) Mediates Non-Transferrin-Bound Iron Uptake into Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617.
- Wang, C.-Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 Is an Iron and Zinc Transporter Whose Cell-Surface Expression Is Up-Regulated by Cellular Iron Loading*. J. Biol. Chem. 2012, 287, 34032–34043.
- Garrick, L.M.; Dolan, K.G.; Romano, M.A.; Garrick, M.D. Non-Transferrin-Bound Iron Uptake in Belgrade and Normal Rat Erythroid Cells. J. Cell. Physiol. 1999, 178, 349–358.
- Tsushima, R.G.; Wickenden, A.D.; Bouchard, R.A.; Oudit, G.Y.; Liu, P.P.; Backx, P.H. Modulation of Iron Uptake in Heart by L-Type Ca2+ Channel Modifiers. Circ. Res. 1999, 84, 1302–1309.
- Kumfu, S.; Chattipakorn, S.; Srichairatanakool, S.; Settakorn, J.; Fucharoen, S.; Chattipakorn, N. T-Type Calcium Channel as a Portal of Iron Uptake into Cardiomyocytes of Beta-Thalassemic Mice. Eur. J. Haematol. 2011, 86, 156–166.
- Lopin, K.V.; Gray, I.P.; Obejero-Paz, C.A.; Thévenod, F.; Jones, S.W. Fe2+ Block and Permeation of CaV3.1 (A1G) T-Type Calcium Channels: Candidate Mechanism for Non–Transferrin-Mediated Fe2+ Influx. Mol. Pharmacol. 2012, 82, 1194–1204.
- Knutson, M.D. Non-Transferrin-Bound Iron Transporters. Free Radic. Biol. Med. 2019, 133, 101–111.
- Liu, Q.; Barker, S.; Knutson, M.D. Iron and Manganese Transport in Mammalian Systems. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118890.
- Ali, M.Y.; Oliva, C.R.; Flor, S.; Griguer, C.E. Mitoferrin, Cellular and Mitochondrial Iron Homeostasis. Cells 2022, 11, 3464.
- Besecker, B.; Bao, S.; Bohacova, B.; Papp, A.; Sadee, W.; Knoell, D.L. The Human Zinc Transporter SLC39A8 (Zip8) Is Critical in Zinc-Mediated Cytoprotection in Lung Epithelia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 294, L1127–L1136.
- Wolff, N.A.; Garrick, L.M.; Zhao, L.; Garrick, M.D.; Thévenod, F. Mitochondria Represent Another Locale for the Divalent Metal Transporter 1 (DMT1). Channels 2014, 8, 458–466.
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thévenod, F. Evidence for Mitochondrial Localization of Divalent Metal Transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145.
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A Role for Divalent Metal Transporter (DMT1) in Mitochondrial Uptake of Iron and Manganese. Sci. Rep. 2018, 8, 211.
- Melino, G.; Stefanini, S.; Chiancone, E.; Antonini, E. Stoichiometry of Iron Oxidation by Apoferritin. FEBS Lett. 1978, 86, 136–138.
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The New Role of Poly (RC)-Binding Proteins as Iron Transport Chaperones: Proteins That Could Couple with Inter-Organelle Interactions to Safely Traffic Iron. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129685.
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each Member of the Poly-r(C)-Binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin*. J. Biol. Chem. 2013, 288, 17791–17802.
- Wang, J.; Pantopoulos, K. Regulation of Cellular Iron Metabolism. Biochem. J. 2011, 434, 365–381.
- Baringer, S.L.; Palsa, K.; Spiegelman, V.S.; Simpson, I.A.; Connor, J.R. Apo- and Holo-Transferrin Differentially Interact with Hephaestin and Ferroportin in a Novel Mechanism of Cellular Iron Release Regulation. J. Biomed. Sci. 2023, 30, 36.
- Pietrangelo, A. Iron and the Liver. Liver Int. 2016, 36 (Suppl. S1), 116–123.
- Camaschella, C. Iron-Deficiency Anemia. N. Engl. J. Med. 2015, 372, 1832–1843.
- Zumerle, S.; Mathieu, J.R.R.; Delga, S.; Heinis, M.; Viatte, L.; Vaulont, S.; Peyssonnaux, C. Targeted Disruption of Hepcidin in the Liver Recapitulates the Hemochromatotic Phenotype. Blood 2014, 123, 3646–3650.
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361.
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-Function Analysis of Ferroportin Defines the Binding Site and an Alternative Mechanism of Action of Hepcidin. Blood 2018, 131, 899–910.
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093.
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787.
- Ward, D.M.; Kaplan, J. Ferroportin-Mediated Iron Transport: Expression and Regulation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1426–1433.
- Wallace, D.F.; Harris, J.M.; Subramaniam, V.N. Functional Analysis and Theoretical Modeling of Ferroportin Reveals Clustering of Mutations According to Phenotype. Am. J. Physiol.-Cell Physiol. 2010, 298, C75–C84.
- Le Gac, G.; Ka, C.; Joubrel, R.; Gourlaouen, I.; Lehn, P.; Mornon, J.-P.; Férec, C.; Callebaut, I. Structure-Function Analysis of the Human Ferroportin Iron Exporter (SLC40A1): Effect of Hemochromatosis Type 4 Disease Mutations and Identification of Critical Residues. Hum. Mutat. 2013, 34, 1371–1380.
- Bonaccorsi Di Patti, M.C.; Polticelli, F.; Cece, G.; Cutone, A.; Felici, F.; Persichini, T.; Musci, G. A Structural Model of Human Ferroportin and of Its Iron Binding Site. FEBS J. 2014, 281, 2851–2860.
- Pao, S.S.; Paulsen, I.T.; Saier, M.H. Major Facilitator Superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34.
- Quistgaard, E.M.; Löw, C.; Guettou, F.; Nordlund, P. Understanding Transport by the Major Facilitator Superfamily (MFS): Structures Pave the Way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132.
- Yan, N. Structural Advances for the Major Facilitator Superfamily (MFS) Transporters. Trends Biochem. Sci. 2013, 38, 151–159.
- Law, C.J.; Maloney, P.C.; Wang, D.-N. Ins and Outs of Major Facilitator Superfamily Antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305.
- Pan, Y.; Ren, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.; Zhang, H.; Fan, X.; et al. Structural Basis of Ion Transport and Inhibition in Ferroportin. Nat. Commun. 2020, 11, 5686.
- Li, S.; Yang, Y.; Li, W. Human Ferroportin Mediates Proton-Coupled Active Transport of Iron. Blood Adv. 2020, 4, 4758–4768.
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of Hepcidin-Bound Ferroportin Reveals Iron Homeostatic Mechanisms. Nature 2020, 586, 807–811.
- Shen, J.; Wilbon, A.S.; Zhou, M.; Pan, Y. Mechanism of Ca2+ Transport by Ferroportin. eLife 2023, 12, e82947.
- Pietrangelo, A. Ferroportin Disease: Pathogenesis, Diagnosis and Treatment. Haematologica 2017, 102, 1972–1984.
- Vlasveld, L.T.; Janssen, R.; Bardou-Jacquet, E.; Venselaar, H.; Hamdi-Roze, H.; Drakesmith, H.; Swinkels, D.W. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals 2019, 12, 132.
- Majore, S.; Bonaccorsi di Patti, M.C.; Valiante, M.; Polticelli, F.; Cortese, A.; Di Bartolomeo, S.; De Bernardo, C.; De Muro, M.; Faienza, F.; Radio, F.C.; et al. Characterization of Three Novel Pathogenic SLC40A1 Mutations and Genotype/Phenotype Correlations in 7 Italian Families with Type 4 Hereditary Hemochromatosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 464–470.
- Ka, C.; Guellec, J.; Pepermans, X.; Kannengiesser, C.; Ged, C.; Wuyts, W.; Cassiman, D.; de Ledinghen, V.; Varet, B.; de Kerguenec, C.; et al. The SLC40A1 R178Q Mutation Is a Recurrent Cause of Hemochromatosis and Is Associated with a Novel Pathogenic Mechanism. Haematologica 2018, 103, 1796–1805.
- Tortosa, V.; di Patti, M.C.B.; Brandi, V.; Musci, G.; Polticelli, F. An Improved Structural Model of the Human Iron Exporter Ferroportin. Insight into the Role of Pathogenic Mutations in Hereditary Hemochromatosis Type 4. Bio-Algorithms Med-Syst. 2017, 13, 215–222.
- Honma, Y.; Karasuyama, T.; Kumamoto, K.; Shimajiri, S.; Toki, Y.; Tatsumi, Y.; Sumida, K.; Koikawa, K.; Morino, K.; Oe, S.; et al. Type 4B Hereditary Hemochromatosis Due to Heterozygous p.D157A Mutation in SLC40A1 Complicated with Hypopituitarism. Med. Mol. Morphol. 2021, 54, 60–67.
- Jiang, L.; Wang, J.; Wang, K.; Wang, H.; Wu, Q.; Yang, C.; Yu, Y.; Ni, P.; Zhong, Y.; Song, Z.; et al. RNF217 Regulates Iron Homeostasis through Its E3 Ubiquitin Ligase Activity by Modulating Ferroportin Degradation. Blood 2021, 138, 689–705.
- Traeger, L.; Wiegand, S.B.; Sauer, A.J.; Corman, B.H.P.; Peneyra, K.M.; Wunderer, F.; Fischbach, A.; Bagchi, A.; Malhotra, R.; Zapol, W.M.; et al. UBA6 and NDFIP1 Regulate the Degradation of Ferroportin. Hematology 2021, 107, 478–488.
- Bayele, H.K.; Srai, S.K.S. A Disease-Causing Mutation K240E Disrupts Ferroportin Trafficking by SUMO (Ferroportin SUMOylation). Biochem. Biophys. Rep. 2021, 25, 100873.
- Yanatori, I.; Richardson, D.R.; Imada, K.; Kishi, F. Iron Export through the Transporter Ferroportin 1 Is Modulated by the Iron Chaperone PCBP2. J. Biol. Chem. 2016, 291, 17303–17318.
- Manatschal, C.; Dutzler, R. The Structural Basis for Metal Ion Transport in the SLC11/NRAMP Family. Chimia 2022, 76, 1005.
- Montalbetti, N.; Simonin, A.; Kovacs, G.; Hediger, M.A. Mammalian Iron Transporters: Families SLC11 and SLC40. Mol. Asp. Med. 2013, 34, 270–287.
- Illing, A.C.; Shawki, A.; Cunningham, C.L.; Mackenzie, B. Substrate Profile and Metal-Ion Selectivity of Human Divalent Metal-Ion Transporter-1. J. Biol. Chem. 2012, 287, 30485–30496.
- Ehrnstorfer, I.A.; Geertsma, E.R.; Pardon, E.; Steyaert, J.; Dutzler, R. Crystal Structure of a SLC11 (NRAMP) Transporter Reveals the Basis for Transition-Metal Ion Transport. Nat. Struct. Mol. Biol. 2014, 21, 990–996.
- Bozzi, A.T.; Bane, L.B.; Weihofen, W.A.; Singharoy, A.; Guillen, E.R.; Ploegh, H.L.; Schulten, K.; Gaudet, R. Crystal Structure and Conformational Change Mechanism of a Bacterial Nramp-Family Divalent Metal Transporter. Structure 2016, 24, 2102–2114.
- Bozzi, A.T.; Bane, L.B.; Zimanyi, C.M.; Gaudet, R. Unique Structural Features in an Nramp Metal Transporter Impart Substrate-Specific Proton Cotransport and a Kinetic Bias to Favor Import. J. Gen. Physiol. 2019, 151, 1413–1429.
- Ehrnstorfer, I.A.; Manatschal, C.; Arnold, F.M.; Laederach, J.; Dutzler, R. Structural and Mechanistic Basis of Proton-Coupled Metal Ion Transport in the SLC11/NRAMP Family. Nat. Commun. 2017, 8, 14033.
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal Structure of a Bacterial Homologue of Na+/Cl--Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223.
- Ray, S.; Berry, S.P.; Wilson, E.A.; Zhang, C.H.; Shekhar, M.; Singharoy, A.; Gaudet, R. High-Resolution Structures with Bound Mn2+ and Cd2+ Map the Metal Import Pathway in an Nramp Transporter. eLife 2023, 12, e84006.
More