Anti-islet autoantibodies serve as key markers in immune-mediated type 1 diabetes (T1D) and slowly progressive T1D (SPIDDM), also known as latent autoimmune diabetes in adults (LADA). Autoantibodies to insulin (IAA), glutamic acid decarboxylase (GADA), tyrosine phosphatase-like protein IA-2 (IA-2A), and zinc transporter 8 (ZnT8A) are currently employed in the diagnosis, pathological analysis, and prediction of T1D. GADA can also be detected in non-diabetic patients with autoimmune diseases other than T1D and may not necessarily reflect insulitis. Conversely, IA-2A and ZnT8A serve as surrogate markers of pancreatic β-cell destruction. A combinatorial analysis of these four anti-islet autoantibodies demonstrated that 93–96% of acute-onset T1D and SPIDDM cases were diagnosed as immune-mediated T1D, while the majority of fulminant T1D cases were autoantibody-negative.
- enzyme-linked immunosorbent assay
- epitope
- glutamic acid decarboxylase
- Latent-Autoimmune Diabetes in Adults (LADA)
- Slowly-progressive type 1 diabetes (SPIDDM)
1. Introduction
2. History of Anti-Islet Autoantibody Discovery
Discovery of islet cell antibodies (ICA) as the first anti-islet autoantibodies in T1D was made by Bottazzo and coworkers in 1974 [5]. ICA detection involved using indirect immunofluorescence on the frozen pancreatic tissue sections of human blood group O, which may recognize various autoantigens. In 1982, Baekkeskov and coworkers discovered autoantibodies against an islet protein with a molecular weight of 64,000 (64 kDa antibody) using the immunoprecipitation method and 35S-methionine-labeled human islet cells [6]. Moreover, in 1983, Palmer and coworkers reported insulin autoantibody (IAA) in insulin naïve new-onset patients with T1D, measured by polyethylene glycol competitive assay using 125I-Tyr A14 human monoiodinated insulin [7]. To overcome the limitation of ICA assay, such as being time-consuming, requiring human pancreatic tissue, and yielding difficulty in obtaining quantitative results, extensive efforts have been made to identify target antigens against ICA using advanced molecular biological techniques such as molecular cloning, gel electrophoresis, polymerase chain reaction, and DNA microarray analysis. To date, more than 10 target antigens have been discovered (Table 1). After identifying the 64kDa islet protein as glutamic acid decarboxylase (GAD) in 1990 [8], several autoantibodies (Figure 1) were discovered. Currently, in addition to IAA and GAD autoantibodies (GADA), tyrosine phosphatase-like protein IA-2 autoantibodies (IA-2A) [9] and zinc transporter 8 autoantibodies (ZnT8A) [10] are employed for the diagnosis, pathological analysis, and prediction of T1D. Additionally, detection methods have evolved from immunohistochemical staining used in ICA to the radioimmunoassay (RIA), enzyme-linked immunosorbent assay (ELISA), and electrochemiluminescence (ECL) assay using recombinant autoantigens produced via prokaryotic and eukaryotic expression systems or in vitro transcription/translation system.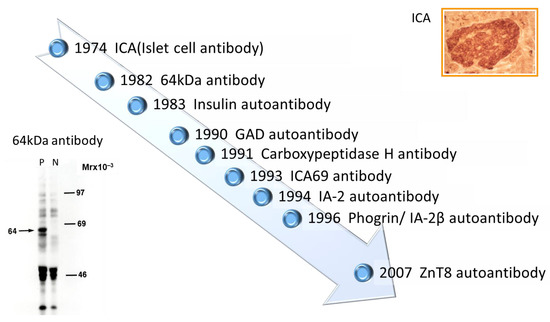
| Name of Antigen | Localization | Function | Reference |
|---|---|---|---|
| Insulin | Insulin secretory granules | Regulate glucose levels in the blood and induce glucose storage in the liver, muscles, and adipose tissue | [7] |
| GAD65 | Synaptic-like vesicles in the cytoplasm of β-cells | Rate-limiting enzyme engaged in the synthesis of the neurotransmitter γ-aminobutyric acid from L-glutamate | [8] |
| GAD67 | Cytosol of β-cells | Rate-limiting enzyme engaged in the synthesis of the neurotransmitter γ-aminobutyric acid from L-glutamate | [11] |
| IA-2 | Insulin secretory granule membrane | Regulate insulin secretory granule content and β-cell growth | [9,12][9][12] |
| Phogrin/IA-2β | Insulin secretory granule membrane | Regulate insulin secretory granule content and β-cell growth | [13,14][13][14] |
| Carboxypeptidase H | Insulin secretory granules and granule membrane | Convert proinsulin into insulin and C-peptide by catalyzing the release of C-terminal arginine or lysine residues from polypeptides | [15] |
| ICA69 | Insulin secretory granule membrane | Dense-core vesicles signaling and maturation | [16] |
| ZnT8 | Insulin secretory granule membrane | Transport zinc ion from the cytosol into the insulin secretory granules | [17,18][17][18] |
| GM2-1 ganglioside | Secretory granules in β-cells and non-β-cells | unknown | [19] |
| Heat shock protein 60 | Insulin secretory granules | Assist correct folding of partially folded polypeptides and presentation of antigen to MHC molecules | [20] |
| GLUT2 | β-cell surface membrane | Uptake glucose from the blood into β-cells | [21] |
| Tetraspanin-7 | Insulin secretory granule membrane | Regulate Ca2+-dependent insulin exocytosis | [22] |
| ICA12/SOX13 | Cytoplasm and nucleus in β-cells and non-β-cells | Transcription factor (Function in the islets is unknown) | [23] |
3. Pathophysiology of the Generation of Anti-Islet Autoantibodies
Although the role of B cells in the pathogenesis of T1D has been extensively studied in non-obese diabetic (NOD) mice, an animal model of human T1D, the mechanism of anti-islet autoantibody generation in humans remains largely unexplored and is currently unknown. However, it is assumed that genetic background, such as MHC class II gene, protein tyrosine phosphatase type 22 gene, and interleukin-2 receptor α gene, and environmental factors are associated with the anti-islet autoantibody generation [24]. In the NOD mice, it has been reported that the escape of islet-specific T cells into the periphery reduced regulatory T cell number and function, and the increased production of autoreactive B cells are thought to play key roles in anti-islet autoantibody generation [24,25][24][25]. Pinto and coworkers reported that increased numbers of thymic B cells and the formation of thymic germinal centers lead to a substantial increase in intrathymic autoantibody levels, resulting in the loss of certain medullary thymic epithelial cells, a decreased negative selection of autoreactive T cells, and an enhanced survival of insulin-reactive thymocytes [26].4. Localization and Function of Autoantigens against Anti-Islet Autoantibodies
- (1)
-
Insulin
- (2)
-
GAD 65 and GAD67
- (3)
-
IA-2 and IA-2β/phogrin
- (4)
-
ZnT8
- (5)
-
Carboxypeptidase H
- (6)
-
ICA69
- (7)
-
GM2-1 ganglioside
- (8)
-
Heat shock protein 60
- (9)
-
GLUT2
- (10)
-
Tetraspanin-7
- (11)
-
ICA12/SOX13
5. Significance of Anti-Islet Autoantibodies in the Pathophysiology of T1D
The Juvenile Diabetes Research Foundation (JDRF), American Diabetes Association (ADA), and Endocrine Society recommend classifying the natural history of T1D into three stages [27] (Figure 2). In Stage 1, autoimmunity to pancreatic islet cells is induced by cytotoxic T cells, and the destruction of pancreatic β-cells (insulitis) begins, even though blood glucose levels remain within the normal range. Two or more IAA, GADA, IA-2A, and ZnT8A, emerge in this stage, with the five-year and ten-year risks of T1D development being approximately 44% and 70%, respectively, while the lifetime risk approaches 100% [28]. However, it is currently understood that anti-islet autoantibodies do not directly cause pancreatic β-cell destruction; they arise as the result of β-cell destruction mediated by T-cells. In Stage 2, similar to Stage 1, individuals test positive for two or more anti-islet autoantibodies, and β-cell destruction progresses, leading to glucose intolerance or dysglycemia. The five-year risk of T1D development at this stage is approximately 75%, and the lifetime risk approaches 100% [29]. Stage 3 is marked by the manifestations of typical clinical symptoms and signs of T1D, including polyuria, polydipsia, weight loss, general fatigue, diabetic ketoacidosis, and others. In this stage, the amount of residual pancreatic β-cells decreases to 20–30% of normal levels. Since anti-islet autoantibodies appear in the peripheral blood during Stage 1 and Stage 2, they are used as a tool for predicting the onset of T1D. The number of positive autoantibodies is more important for prediction than the specific positive autoantibodies [30].Additionally, a study involving first-degree relatives of patients with T1D reported that IAA or GADA appeared first, followed by IA-2A and ZnT8A directly before the onset of diabetes [31]. We observed a similar sequential emergence of anti-islet autoantibodies in patients with T1D who developed the condition during interferon therapy [32]. Based on this evidence, IA-2A and ZnT8A are considered surrogate markers for pancreatic β-cell destruction. This is supported by a report demonstrating the epitope spreading of IA-2A during the preclinical stage of T1D [33]. Conversely, as shown in Table 2, GADA is detected not only in patients with acute-onset T1D and SPIDDM, but also in some patients with fulminant T1D and non-diabetic patients with polyglandular autoimmune syndrome, autoimmune thyroid disease (AITD), and stiff-person syndrome. Therefore, unlike IA-2A and ZnT8A, it is presumed that GADA may not be a specific marker for pancreatic β-cell destruction. In our case, where GADA became re-elevated more than 10 years after the onset of T1D, anti-thyroid autoantibodies turned positive directly before the re-elevation of GADA [34]. Therefore, it is important to note that GADA may be associated with thyroid autoimmunity rather than insulitis in some instances.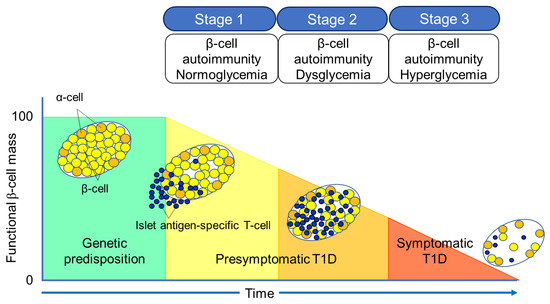 Figure 2. Three stages of natural history of T1D.Table 2. Disease specificity of GAD autoantibodies.
Figure 2. Three stages of natural history of T1D.Table 2. Disease specificity of GAD autoantibodies.Subject Prevalence Healthy control <1% Acute-onset type 1 diabetes (at onset) 60–80% Fulminant type 1 diabetes 5–9% LADA (SPIDDM) 100% Type 2 diabetes (diet/OHA) 4–5% Polyglandular autoimmune syndrome, type 1 30–40% Polyglandular autoimmune syndrome, type 2 30–50% Autoimmune thyroid disease 6–8% Stiff-person syndrome 60–70% LADA, latent-autoimmune diabetes in adults; SPIDDM, slowly progressive insulin-dependent diabetes; OHA, oral hypoglycemic agents.6. Role of Anti-Islet Autoantibodies in the Diagnosis of T1D
In this section, we discuss the key considerations when diagnosing T1D using anti-islet autoantibodies. First, because distinguishing IAA from insulin antibodies produced by exogenous insulin injection is challenging, we considered patients positive for IAA if antibodies to insulin were present in insulin-naïve patients or those within 2 weeks of initiating insulin therapy. Our study using sera obtained within 2 weeks after onset revealed that the prevalence of IAA, GADA, IA-2A, and ZnT8A in patients with acute-onset T1D was 58, 80, 66, and 63%, respectively (Figure 3A). Similarly, the prevalence of these autoantibodies in SPIDDM patients was 41, 83, 27, and 27%, respectively (Figure 3B). Thus, a combinatorial analysis of the four autoantibodies indicated that 96% of acute-onset T1D and 93% of SPIDDM were immune-mediated T1D, while approximately 5% were classified as idiopathic T1D [35]. These data are consistent with previous reports from studies conducted in countries with Caucasoid populations [10,36,37,38][10][36][37][38]. In contrast, the majority of patients with fulminant T1D were negative for anti-islet autoantibodies (Figure 3C) and are classified into idiopathic T1D, suggesting that the mechanism of β-cell destruction in fulminant T1D may differ from acute-onset T1D and SPIDDM.In Japan, medical insurance guidelines instruct physicians to measure GADA first when T1D is suspected. However, about 10% of non-fulminant T1D patients are positive for other anti-islet autoantibodies without GADA. Therefore, measuring multiple autoantibodies is crucial for the accurate diagnosis of immune-mediated T1D.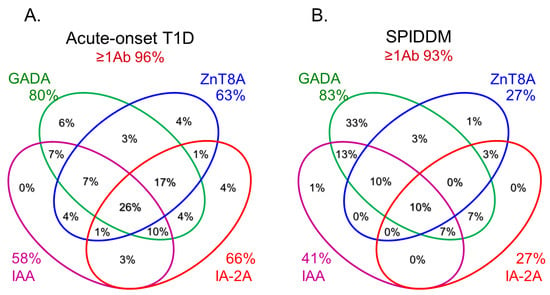
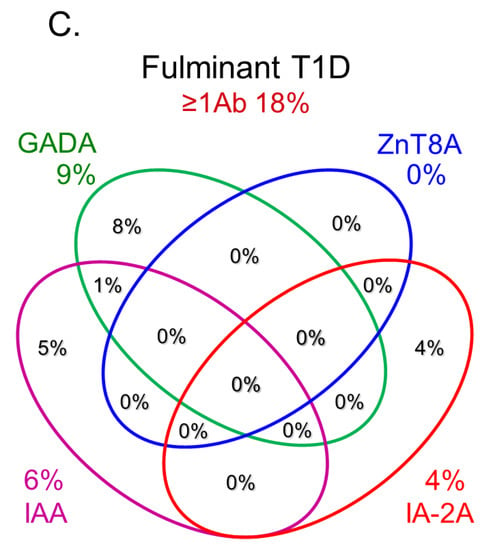 Figure 3.Combinatorial analysis of anti-islet autoantibodies in patients with acute-onset T1D (A), SPIDDM (B), and fulminant T1D (C
Figure 3.Combinatorial analysis of anti-islet autoantibodies in patients with acute-onset T1D (A), SPIDDM (B), and fulminant T1D (CReferences
- ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, Collins BS, Hilliard ME, Isaacs D, Johnson EL, Kahan S, Khunti K, Leon J, Lyons SK, Perry ML, Prahalad P, Pratley RE, Seley JJ, Stanton RC, Gabbay RA, on behalf of the American Diabetes Association. Classification and diagnosis of diabetes: Standards of care in diabetes-2023. Diabetes Care 46(Supplement_1): S19-S40, 2023
- Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat Rev Endocrinol. 17:150-161, 2021
- Karvonen M, Viik-Kajander M, Moltchanova E, Libman I, LaPorte R, Tuomilehto J. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care. 23:1516-26, 2000
- Kawasaki E, Matsuura N, Eguchi K. Type 1 diabetes in Japan. Diabetologia. 49:828-836, 2006
- Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet. 2(7892):1279-1283, 1974
- Baekkeskov S, Nielsen JH, Marner B, Bilde T, Ludvigsson J, Lernmark Å. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature. 298(5870):167-169, 1982
- Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science. 222(4630):1337-9, 1983
- Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, De Camilli P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 347(6289):151-156, 1990
- Christgau S, Schierbeck H, Aanstoot HJ, Aagaard L, Begley K, Kofod H, Hejnaes K, Baekkeskov S. Pancreatic beta cells express two autoantigenic forms of glutamic acid decarboxylase, a 65-kDa hydrophilic form and a 64-kDa amphiphilic form which can be both membrane-bound and soluble. J Biol Chem. 266:21257-21264, 1991
- Lan MS, Lu J, Goto Y, Notkins AL. Molecular cloning and identification of a receptor-type protein tyrosine phosphatase, IA-2, from human insulinoma. DNA Cell Biol. 13:505-14, 1994
- Harashima S, Clark A, Christie MR, Notkins AL. The dense core transmembrane vesicle protein IA-2 is a regulator of vesicle number and insulin secretion. Proc Natl Acad Sci USA. 102:8704-8709, 2005
- Kawasaki E, Hutton JC, Eisenbarth GS. Molecular cloning and characterization of the human transmembrane protein tyrosine phosphatase homologue, phogrin, an autoantigen of type 1 diabetes. Biochem Biophys Res Commun. 227:440-447, 1996
- Cai T, Hirai H, Zhang G, Zhang M, Takahashi N, Kasai H, Satin LS, Leapman RD, Notkins AL. Deletion of IA-2 and/or IA-2β in mice decreases insulin secretion by reducing the number of dense core vesicles. Diabetologia. 54:2347-2357, 2011
- Aguilar-Diosdado M, Parkinson D, Corbett JA, Kwon G, Marshall CA, Gingerich RL, Santiago JV, McDaniel ML. Potential autoantigens in IDDM. Expression of carboxypeptidase-H and insulin but not glutamate decarboxylase on the β-cell surface. Diabetes. 43:418-425, 1994
- Spitzenberger F, Pietropaolo S, Verkade P, Habermann B, Lacas-Gervais S, Mziaut H, Pietropaolo M, Solimena M. Islet cell autoantigen of 69 kDa is an arfaptin-related protein associated with the Golgi complex of insulinoma INS-1 cells. J Biol Chem. 278:26166-26173, 2003
- Dwivedi OP, Lehtovirta M, Hastoy B, Chandra V, Krentz NAJ, Kleiner S, Jain D, Richard AM, Abaitua F, Beer NL, Grotz A, Prasad RB, Hansson O, Ahlqvist E, Krus U, Artner I, Suoranta A, Gomez D, Baras A, Champon B, Payne AJ, Moralli D, Thomsen SK, Kramer P, Spiliotis I, Ramracheya R, Chabosseau P, Theodoulou A, Cheung R, van de Bunt M, Flannick J, Trombetta M, Bonora E, Wolheim CB, Sarelin L, Bonadonna RC, Rorsman P, Davies B, Brosnan J, McCarthy MI, Otonkoski T, Lagerstedt JO, Rutter GA, Gromada J, Gloyn AL, Tuomi T, Groop L. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet 51, 1596-1606, 2019
- Nicolson TJ, Bellomo EA, Wijesekara N, Loder MK, Baldwin JM, Gyulkhandanyan AV, Koshkin V, Tarasov AI, Carzaniga R, Kronenberger K, Taneja TK, da Silva Xavier G, Libert S, Froguel P, Scharfmann R, Stetsyuk V, Ravassard P, Parker H, Gribble FM, Reimann F, Sladek R, Hughes SJ, Johnson PR, Masseboeuf M, Burcelin R, Baldwin SA, Liu M, Lara-Lemus R, Arvan P, Schuit FC, Wheeler MB, Chimienti F, Rutter GA. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 58, 2070-2083, 2009
- Dotta F, Previti M, Neerman-Arbez M, Dionisi S, Cucinotta D, Lenti L, Di Mario U, Halban PA. The GM2-1 ganglioside islet autoantigen in insulin-dependent diabetes mellitus is expressed in secretory granules and is not β-cell specific. Endocrinology. 139:316-319, 1998
- Ozawa Y, Kasuga A, Nomaguchi H, Maruyama T, Kasatani T, Shimada A, Takei I, Miyazaki J, Saruta T. Detection of autoantibodies to the pancreatic islet heat shock protein 60 in insulin-dependent diabetes mellitus. J Autoimmun. 9:517-524, 1996
- Inman LR, McAllister CT, Chen L, Hughes S, Newgard CB, Kettman JR, Unger RH, Johnson JH. Autoantibodies to the GLUT-2 glucose transporter of β cells in insulin-dependent diabetes mellitus of recent onset. Proc Natl Acad Sci USA. 90:1281-1284, 1993
- Walther D, Eugster A, Jergens S, Gavrisan A, Weinzierl C, Telieps T, Winkler C, Ziegler AG, Bonifacio E. Tetraspanin 7 autoantibodies in type 1 diabetes. Diabetologia. 59:1973-1976, 2016
- Kasimiotis H, Myers MA, Argentaro A, Mertin S, Fida S, Ferraro T, Olsson J, Rowley MJ, Harley VR. Sex-determining region Y-related protein SOX13 is a diabetes autoantigen expressed in pancreatic islets. Diabetes. 49:555-561, 2000
- Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, Rewers M, Eisenbarth GS, Jensen J, Davidson HW, Hutton JC. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA. 104:17040-17045, 2007
- Fousteri G, Ippolitoa E, Ahmedb R, Hamad ARA. Beta-cell specific autoantibodies: Are they just an indicator of type 1 diabetes? Curr Diabetes Rev. 13: 322-329, 2017
- Mallone R, Brezar V. To B or not to B: (anti)bodies of evidence on the crime scene of type 1 diabetes? Diabetes 60:2020-2022, 2011
- Pinto AI, Smith J, Kissack MR, Hogg KG, Green EA. Thymic B cell-mediated attack of thymic stroma precedes type 1 diabetes development. Front Immunol 9:1281, 2018
- Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, Greenbaum CJ, Herold KC, Krischer JP, Lernmark Å, Ratner RE, Rewers MJ, Schatz DA, Skyler JS, Sosenko JM, Ziegler AG. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. 38:1964-1974, 2015
- Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, Bonifacio E, Eisenbarth GS. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309: 2473-2479, 2013
- Krischer JP; Type 1 Diabetes TrialNet Study Group. The use of intermediate endpoints in the design of type 1 diabetes prevention trials. Diabetologia 56: 1919-1924, 2013
- Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS.Prediction of type 1 diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 45:926-933, 1996
- Yu L, Rewers M, Gianani R, Kawasaki E, Zhang Y, Verge C, Chase P, Klingensmith G, Erlich H, Norris J, Eisenbarth GS. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J Clin Endocrinol Metab. 81(12):4264-4267, 1996
- Nakamura K, Kawasaki E, Abiru N, Jo O, Fukushima K, Satoh T, Kuriya G, Kobayashi M, Kuwahara H, Yamasaki H, Ide T, Eguchi K. Trajectories of anti-islet autoantibodies before development of type 1 diabetes in interferon-treated hepatitis C patients. Case reports and a literature review. Endocr J. 57:947-951, 2010
- Kawasaki E, Yu L, Rewers MJ, Hutton JC, Eisenbarth GS. Definition of multiple ICA512/phogrin autoantibody epitopes and detection of intramolecular epitope spreading in relatives of patients with type 1 diabetes. Diabetes. 47:733-742, 1998
- Kawasaki E, Yasui J, Tsurumaru M, Takashima H, Ikeoka T, Mori F, Akazawa S, Ueki I, Kobayashi M, Kuwahara H, Abiru N, Yamasaki H, Kawakami A. Sequential elevation of autoantibodies to thyroglobulin and glutamic acid decarboxylase in type 1 diabetes. World J Diabetes. 4:227-30, 2013
- Kawasaki E, Nakamura K, Kuriya G, Satoh T, Kobayashi M, Kuwahara H, Abiru N, Yamasaki H, Matsuura N, Miura J, Uchigata Y, Eguchi K.Differences in the humoral autoreactivity to zinc transporter 8 between childhood- and adult-onset type 1 diabetes in Japanese patients. Clin Immunol. 138:146-153, 2011
References
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. Classification and diagnosis of diabetes: Standards of care in diabetes-2023. Diabetes Care 2023, 46 (Suppl. S1), S19–S40.
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2021, 17, 150–161.
- Karvonen, M.; Viik-Kajander, M.; Moltchanova, E.; Libman, I.; LaPorte, R.; Tuomilehto, J. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care 2000, 23, 1516–1526.
- Kawasaki, E.; Matsuura, N.; Eguchi, K. Type 1 diabetes in Japan. Diabetologia 2006, 49, 828–836.
- Bottazzo, G.F.; Florin-Christensen, A.; Doniach, D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974, 2, 1279–1283.
- Baekkeskov, S.; Nielsen, J.H.; Marner, B.; Bilde, T.; Ludvigsson, J.; Lernmark, Å. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature 1982, 298, 167–169.
- Palmer, J.P.; Asplin, C.M.; Clemons, P.; Lyen, K.; Tatpati, O.; Raghu, P.K.; Paquette, T.L. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 1983, 222, 1337–1339.
- Baekkeskov, S.; Aanstoot, H.J.; Christgau, S.; Reetz, A.; Solimena, M.; Cascalho, M.; Folli, F.; Richter-Olesen, H.; De Camilli, P. Identification of the 64 K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 1990, 347, 151–156.
- Lan, M.S.; Lu, J.; Goto, Y.; Notkins, A.L. Molecular cloning and identification of a receptor-type protein tyrosine phosphatase, IA-2, from human insulinoma. DNA Cell. Biol. 1994, 13, 505–514.
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 17040–17045.
- Christgau, S.; Schierbeck, H.; Aanstoot, H.J.; Aagaard, L.; Begley, K.; Kofod, H.; Hejnaes, K.; Baekkeskov, S. Pancreatic beta cells express two autoantigenic forms of glutamic acid decarboxylase, a 65-kDa hydrophilic form and a 64-kDa amphiphilic form which can be both membrane-bound and soluble. J. Biol. Chem. 1991, 266, 21257–21264.
- Harashima, S.; Clark, A.; Christie, M.R.; Notkins, A.L. The dense core transmembrane vesicle protein IA-2 is a regulator of vesicle number and insulin secretion. Proc. Natl. Acad. Sci. USA 2005, 102, 8704–8709.
- Kawasaki, E.; Hutton, J.C.; Eisenbarth, G.S. Molecular cloning and characterization of the human transmembrane protein tyrosine phosphatase homologue, phogrin, an autoantigen of type 1 diabetes. Biochem. Biophys. Res. Commun. 1996, 227, 440–447.
- Cai, T.; Hirai, H.; Zhang, G.; Zhang, M.; Takahashi, N.; Kasai, H.; Satin, L.S.; Leapman, R.D.; Notkins, A.L. Deletion of IA-2 and/or IA-2β in mice decreases insulin secretion by reducing the number of dense core vesicles. Diabetologia 2011, 54, 2347–2357.
- Aguilar-Diosdado, M.; Parkinson, D.; Corbett, J.A.; Kwon, G.; Marshall, C.A.; Gingerich, R.L.; Santiago, J.V.; McDaniel, M.L. Potential autoantigens in IDDM. Expression of carboxypeptidase-H and insulin but not glutamate decarboxylase on the β-cell surface. Diabetes 1994, 43, 418–425.
- Spitzenberger, F.; Pietropaolo, S.; Verkade, P.; Habermann, B.; Lacas-Gervais, S.; Mziaut, H.; Pietropaolo, M.; Solimena, M. Islet cell autoantigen of 69 kDa is an arfaptin-related protein associated with the Golgi complex of insulinoma INS-1 cells. J. Biol. Chem. 2003, 278, 26166–26173.
- Dwivedi, O.P.; Lehtovirta, M.; Hastoy, B.; Chandra, V.; Krentz, N.A.J.; Kleiner, S.; Jain, D.; Richard, A.M.; Abaitua, F.; Beer, N.L.; et al. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat. Genet. 2019, 51, 1596–1606.
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K.; et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58, 2070–2083.
- Dotta, F.; Previti, M.; Neerman-Arbez, M.; Dionisi, S.; Cucinotta, D.; Lenti, L.; Di Mario, U.; Halban, P.A. The GM2-1 ganglioside islet autoantigen in insulin-dependent diabetes mellitus is expressed in secretory granules and is not β-cell specific. Endocrinology 1998, 139, 316–319.
- Ozawa, Y.; Kasuga, A.; Nomaguchi, H.; Maruyama, T.; Kasatani, T.; Shimada, A.; Takei, I.; Miyazaki, J.; Saruta, T. Detection of autoantibodies to the pancreatic islet heat shock protein 60 in insulin-dependent diabetes mellitus. J. Autoimmun. 1996, 9, 517–524.
- Inman, L.R.; McAllister, C.T.; Chen, L.; Hughes, S.; Newgard, C.B.; Kettman, J.R.; Unger, R.H.; Johnson, J.H. Autoantibodies to the GLUT-2 glucose transporter of β cells in insulin-dependent diabetes mellitus of recent onset. Proc. Natl. Acad. Sci. USA 1993, 90, 1281–1284.
- Walther, D.; Eugster, A.; Jergens, S.; Gavrisan, A.; Weinzierl, C.; Telieps, T.; Winkler, C.; Ziegler, A.G.; Bonifacio, E. Tetraspanin 7 autoantibodies in type 1 diabetes. Diabetologia 2016, 59, 1973–1976.
- Kasimiotis, H.; Myers, M.A.; Argentaro, A.; Mertin, S.; Fida, S.; Ferraro, T.; Olsson, J.; Rowley, M.J.; Harley, V.R. Sex-determining region Y-related protein SOX13 is a diabetes autoantigen expressed in pancreatic islets. Diabetes 2000, 49, 555–561.
- Fousteri, G.; Ippolitoa, E.; Ahmedb, R.; Hamad, A.R.A. Beta-cell specific autoantibodies: Are they just an indicator of type 1 diabetes? Curr. Diabetes Rev. 2017, 13, 322–329.
- Mallone, R.; Brezar, V. To B or not to B: (anti)bodies of evidence on the crime scene of type 1 diabetes? Diabetes 2011, 60, 2020–2022.
- Pinto, A.I.; Smith, J.; Kissack, M.R.; Hogg, K.G.; Green, E.A. Thymic B cell-mediated attack of thymic stroma precedes type 1 diabetes development. Front. Immunol. 2018, 9, 1281.
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974.
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479.
- Krischer, J.P. Type 1 Diabetes TrialNet Study Group. The use of intermediate endpoints in the design of type 1 diabetes prevention trials. Diabetologia 2013, 56, 1919–1924.
- Verge, C.F.; Gianani, R.; Kawasaki, E.; Yu, L.; Pietropaolo, M.; Jackson, R.A.; Chase, H.P.; Eisenbarth, G.S. Prediction of type 1 diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996, 45, 926–933.
- Yu, L.; Rewers, M.; Gianani, R.; Kawasaki, E.; Zhang, Y.; Verge, C.; Chase, P.; Klingensmith, G.; Erlich, H.; Norris, J.; et al. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J. Clin. Endocrinol. Metab. 1996, 81, 4264–4267.
- Nakamura, K.; Kawasaki, E.; Abiru, N.; Jo, O.; Fukushima, K.; Satoh, T.; Kuriya, G.; Kobayashi, M.; Kuwahara, H.; Yamasaki, H.; et al. Trajectories of anti-islet autoantibodies before development of type 1 diabetes in interferon-treated hepatitis C patients. Case reports and a literature review. Endocr. J. 2010, 57, 947–951.
- Kawasaki, E.; Yu, L.; Rewers, M.J.; Hutton, J.C.; Eisenbarth, G.S. Definition of multiple ICA512/phogrin autoantibody epitopes and detection of intramolecular epitope spreading in relatives of patients with type 1 diabetes. Diabetes 1998, 47, 733–742.
- Kawasaki, E.; Yasui, J.; Tsurumaru, M.; Takashima, H.; Ikeoka, T.; Mori, F.; Akazawa, S.; Ueki, I.; Kobayashi, M.; Kuwahara, H.; et al. Sequential elevation of autoantibodies to thyroglobulin and glutamic acid decarboxylase in type 1 diabetes. World J. Diabetes 2013, 4, 227–230.
- Kawasaki, E.; Nakamura, K.; Kuriya, G.; Satoh, T.; Kobayashi, M.; Kuwahara, H.; Abiru, N.; Yamasaki, H.; Matsuura, N.; Miura, J.; et al. Differences in the humoral autoreactivity to zinc transporter 8 between childhood- and adult-onset type 1 diabetes in Japanese patients. Clin. Immunol. 2011, 138, 146–153.
- Achenbach, P.; Lampasona, V.; Landherr, U.; Koczwara, K.; Krause, S.; Grallert, H.; Winkler, C.; Pflüger, M.; Illig, T.; Bonifacio, E.; et al. Autoantibodies to zinc transporter 8 and SLC30A8 genotype stratify type 1 diabetes risk. Diabetologia 2009, 52, 1881–1888.
- Wenzlau, J.M.; Frisch, L.M.; Hutton, J.C.; Fain, P.R.; Davidson, H.W. Changes in zinc transporter 8 autoantibodies following type 1 diabetes onset: The type 1 diabetes genetics consortium autoantibody workshop. Diabetes Care 2015, 38 (Suppl. S2), S14–S20.
- Vermeulen, I.; Weets, I.; Asanghanwa, M.; Ruige, J.; Van Gaal, L.; Mathieu, C.; Keymeulen, B.; Lampasona, V.; Wenzlau, J.M.; Hutton, J.C.; et al. Contribution of antibodies against IA-2β and zinc transporter 8 to classification of diabetes diagnosed under 40 years of age. Diabetes Care 2011, 34, 1760–1765.