Targeted therapies for solid tumors, including non-small cell lung cancer (NSCLC), have advanced significantly, offering tailored treatment options for patients. However, individuals without targetable mutations pose a clinical challenge, as they may not respond to standard treatments like immune-checkpoint inhibitors (ICIs) and novel targeted therapies; further, there is an unmet need to identify prognostic biomarkers of response to treatment. Inflammatory cytokines such as interleukin-1 beta (IL-1β) have emerged as a key area of focus and hold significant potential implications for future clinical practice. Combinatorial approaches of IL-1β inhibitors and ICIs may provide a potential therapeutic modality for NSCLC patients without targetable mutations and offer insight into the continued search for prognostic biomarkers.
- ICIs
- IL-1β
- therapeutic resistance
- inflammation
- signaling
- NSCLC
- tumor microenvironment
1. Introduction
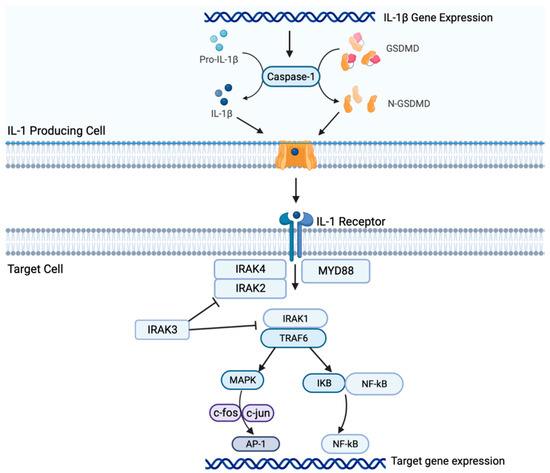
2. Il-1β Signaling in NSCLC
8. Conclusions
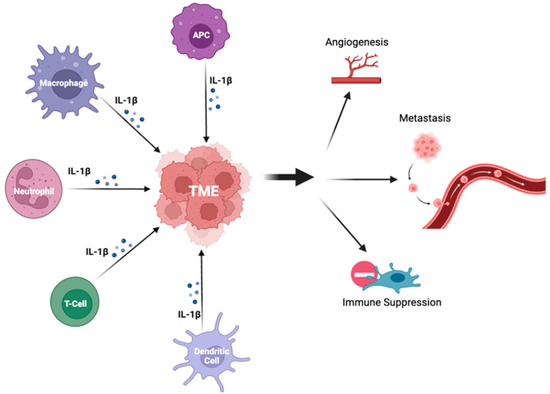
Both preclinical and clinical studies suggest that the IL-1β/PD-1/PD-L1 pathway plays a crucial role in carcinogenesis of NSCLC and interacts within the TME. IL-1β signaling may be associated with resistance to therapy and represents a novel therapeutic target for NSCLC. However, further studies are warranted to fully elucidate the underlying mechanisms and determine the optimal combination regimens. As research into the IL-1β pathway continues to progress, it is hoped that the development of new therapeutic strategies will lead to standardized prediction models and biomarkers of response, and ultimately, improved outcomes for patients with NSCLC.References
- Viviane Teixeira L. de Alencar; Amanda B. Figueiredo; Marcelo Corassa; Kenneth J. Gollob; Vladmir C. Cordeiro de Lima; Lung cancer in never smokers: Tumor immunology and challenges for immunotherapy. Front. Immunol. 2022, 13, 984349.
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009.
- Ming Yi; Mengke Niu; Linping Xu; Suxia Luo; Kongming Wu; Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10.
- Roy S. Herbst; Daniel Morgensztern; Chris Boshoff; The biology and management of non-small cell lung cancer. Nat. 2018, 553, 446-454.
- Xi Yan; Lina Han; Riyang Zhao; Sumaya Fatima; Lianmei Zhao; Feng Gao; Prognosis value of IL-6, IL-8, and IL-1β in serum of patients with lung cancer: A fresh look at interleukins as a biomarker. Heliyon 2022, 8, e09953.
- Cédric Rébé; François Ghiringhelli; Interleukin-1β and Cancer. Cancers 2020, 12, 1791.
- Shuhang Wang; Pei Yuan; Beibei Mao; Ning Li; Jianming Ying; Xiuli Tao; Wei Tang; Lei Zhang; Xiao Geng; Fan Zhang; et al.Qi XueLijia WuHenghui ZhangShugeng GaoJie He Genomic features and tumor immune microenvironment alteration in NSCLC treated with neoadjuvant PD-1 blockade. npj Precis. Oncol. 2022, 6, 1-8.
- Jun Zhang; Nirmal Veeramachaneni; Targeting interleukin-1β and inflammation in lung cancer. Biomark. Res. 2022, 10, 1-9.
- Xiang Qin; Tingting Li; Shun Li; Hong Yang; Chunhui Wu; Chuan Zheng; Fengming You; Yiyao Liu; The tumor biochemical and biophysical microenvironments synergistically contribute to cancer cell malignancy. null 2019, 17, 1186-1187.
- Jay M Lee; Masahiro Tsuboi; Edward S Kim; Tony Sk Mok; Pilar Garrido; Overcoming immunosuppression and pro-tumor inflammation in lung cancer with combined IL-1β and PD-1 inhibition. Futur. Oncol. 2022, 18, 3085-3100.
- Moshe Elkabets; Vera S. G. Ribeiro; Charles A. Dinarello; Suzanne Ostrand-Rosenberg; James P. Di Santo; Ron N. Apte; Christian A. J. Vosshenrich; IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347-3357.
- Shengjun Li; Wei Wang; Ning Zhang; Tingxian Ma; Chenghai Zhao; IL-1β mediates MCP-1 induction by Wnt5a in gastric cancer cells. BMC Cancer 2014, 14, 480-480.
- Ryan Kolb; Liem Phan; Nicholas Borcherding; Yinghong Liu; Fang Yuan; Ann M. Janowski; Qing Xie; Kathleen R. Markan; Wei Li; Matthew J. Potthoff; et al.Enrique Fuentes-MatteiLesley G. ElliesC. Michael KnudsonMong-Hong LeeSai-Ching J. YeungSuzanne L. CasselFayyaz S. SutterwalaWeizhou Zhang Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007.
- Yaron Carmi; Shahar Dotan; Peleg Rider; Irena Kaplanov; Malka R. White; Rona Baron; Shai Abutbul; Monica Huszar; Charles A. Dinarello; Ron N. Apte; et al.Elena Voronov The Role of IL-1β in the Early Tumor Cell–Induced Angiogenic Response. null 2013, 190, 3500-3509.
- Jian Huang; Xiuwen Lan; Ting Wang; Hailing Lu; Mengru Cao; Shi Yan; Yue Cui; Dexin Jia; Li Cai; Ying Xing; et al. Targeting the IL-1β/EHD1/TUBB3 axis overcomes resistance to EGFR-TKI in NSCLC. Oncogene 2019, 39, 1739-1755.
- Erin Fahey; Sarah L. Doyle; IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426.
- Pei Hai Ping; Tan Feng Bo; Liu Li; Yu Nan Hui; Zhu Hong; IL-1β/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch. Biochem. Biophys. 2016, 604, 20-26.
- Chen Wu; Bin Xu; You Zhou; Mei Ji; Dachuan Zhang; Jingting Jiang; Changping Wu; Correlation between serum IL-1β and miR-144-3p as well as their prognostic values in LUAD and LUSC patients. Oncotarget 2016, 7, 85876-85887.
- M. F. Sanmamed; J. L. Perez-Gracia; K. A. Schalper; J. P. Fusco; A. Gonzalez; M. E. Rodriguez-Ruiz; C. Oñate; G. Perez; C. Alfaro; S. Martín-Algarra; et al.M. P. AnduezaA. GurpideM. MorgadoJ. WangA. BacchiocchiR. HalabanH. KlugerL. ChenM. SznolI. Melero Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988-1995.
- Seth B. Coffelt; Kelly Kersten; Chris W. Doornebal; Jorieke Weiden; Kim Vrijland; Cheei-Sing Hau; Niels J. M. Verstegen; Metamia Ciampricotti; Lukas J. A. C. Hawinkels; Jos Jonkers; et al.Karin E. De Visser IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nat. 2015, 522, 345-348.
- Chengcheng Jin; Georgia K. Lagoudas; Chen Zhao; Susan Bullman; Arjun Bhutkar; Bo Hu; Samuel Ameh; Demi Sandel; Xu Sue Liang; Sarah Mazzilli; et al.Mark T. WharyMatthew MeyersonRonald GermainPaul C. BlaineyJames G. FoxTyler Jacks Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998-1013.e16.
- Nicholas L Syn; Michele W L Teng; Tony S K Mok; Ross A Soo; De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731-e741.
- Edward B. Garon; James Chih-Hsin Yang; Steven M. Dubinett; The Role of Interleukin 1β in the Pathogenesis of Lung Cancer. JTO Clin. Res. Rep. 2020, 1, 100001.
- Valerio Gelfo; Donatella Romaniello; Martina Mazzeschi; Michela Sgarzi; Giada Grilli; Alessandra Morselli; Beatrice Manzan; Karim Rihawi; Mattia Lauriola; Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009.
- Ningning Yan; Sanxing Guo; Huixian Zhang; Ziheng Zhang; Shujing Shen; Xingya Li; BRAF-Mutated Non-Small Cell Lung Cancer: Current Treatment Status and Future Perspective. Front. Oncol. 2022, 12, 863043.
- Eva Hajek; Franziska Krebs; Rebekka Bent; Katharina Haas; Antje Bast; Ivo Steinmetz; Andrea Tuettenberg; Stephan Grabbe; Matthias Bros; BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget 2018, 9, 28294-28308.
- Angela M. Davies; Primo N. Lara; Philip C. Mack; Paul H. Gumerlock; Richard J. Bold; David R. Gandara; Bortezomib-Based Combinations in the Treatment of Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2005, 7, S59-S63.
- Allyson G. McLoed; Taylor P. Sherrill; Dong-Sheng Cheng; Wei Han; Jamie A. Saxon; Linda A. Gleaves; Pingsheng Wu; Vasiliy V. Polosukhin; Michael Karin; Fiona E. Yull; et al.Georgios T. StathopoulosVassilis GeorgouliasRinat ZaynagetdinovTimothy S. Blackwell Neutrophil-Derived IL-1β Impairs the Efficacy of NF-κB Inhibitors against Lung Cancer. Cell Rep. 2016, 16, 120-132.
- Yuan, B.; Clowers, M.J.; Velasco, W.V.; Peng, S.; Peng, Q.; Shi, Y.; Ramos-Castaneda, M.; Zarghooni, M.; Yang, S.; Babcock, R.L.; et al.et al. Targeting IL-1β as an immunopreventive and therapeutic modality for K-ras-mutant lung cancer.. JCI Insight 2022, 7, e157788.