Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Chitra Vinnakota and Version 2 by Lindsay Dong.
Glutamate N-methyl-D-aspartate receptor (NMDAR) hypofunction has been proposed to underlie schizophrenia symptoms. This theory arose from the observation that administration of NMDAR antagonists, which are compounds that inhibit NMDAR activity, reproduces behavioural and molecular schizophrenia-like phenotypes, including hallucinations, delusions and cognitive impairments in healthy humans and animal models. However, the role of specific NMDAR subunits in these schizophrenia-relevant phenotypes is largely unknown. Mounting evidence implicates the GluN2D subunit of NMDAR in some of these symptoms and pathology.
- GluN2D
- schizophrenia
- NMDA receptor
- NMDAR antagonists
1. Schizophrenia
Schizophrenia is a severe, debilitating, chronic neuropsychiatric disorder with a lifetime prevalence of 0.72% [1]. It has a complex and heterogeneous presentation making it difficult to diagnose and to identify a consistent underlying aetiology or pathology. People with schizophrenia demonstrate high inter-individual variability with respect to symptoms, disease course, outcome and response to treatment. Schizophrenia symptoms are divided into three main classes: positive, negative and cognitive deficits. Of the three, positive symptoms are the most easily identifiable and are defined as psychotic features that are not usually present in healthy people, and include hallucinations, delusions and disorganised speech and behaviour [2][3][2,3]. Negative symptoms refer to a reduction or disruption in normal emotions and behaviours manifesting as social and emotional withdrawal, apathy and avolition [2][4][2,4]. Cognitive symptoms vary in severity amongst people with schizophrenia and include elements such as deficits in verbal memory, working memory, attention, executive functioning, cognitive flexibility and processing speed [5].
Schizophrenia typically has an early onset, with most people being diagnosed in their late adolescence—early adulthood years. The onset of the first psychotic episode is usually preceded by a prodromal period during which symptoms gradually emerge and this period can last several years. An early onset combined with long-term deficits in social, educational and occupational function make this disorder one of the leading causes of chronic disability with significant impacts on the quality of life of patients and their families and caregivers [6][7][6,7].
The causes of schizophrenia are not fully understood but are thought to be multifactorial, involving a complex interplay between multiple genetic variants and environmental factors. Genome-wide association studies have identified multiple common variants of small effect spanning over 250 genetic loci, suggesting that schizophrenia is a polygenic disorder in most cases [8][9][10,11]. Genes associated with schizophrenia risk are involved in various functions, including the regulation of the postsynaptic membrane, synaptic transmission and neurodevelopmental pathways, including glutamate pathways [9][10][11][11,12,13]. Non-genetic factors that increase lifetime risk for schizophrenia include obstetric complications, advanced paternal age, living in an urban setting, childhood trauma or adversity, cannabis use and first-generation migration [12][13][14][15][16][17][18][19][14,15,16,17,18,19,20,21]. Although there have been advances in our understanding of risk factors associated with schizophrenia, the aetiological complexity has made it a challenge to identify the underlying disease mechanisms and find effective cures and preventative strategies.
Currently available pharmacological treatments, chiefly conventional and atypical anti-psychotics, and psychotherapy, have proven clinical utility and can help manage positive symptoms in some people [20][21][22,23]. However, antipsychotic medications have limited efficacy and are poorly tolerated, with substantial side effects in approximately 30% of people. Moreover, they usually offer little benefit in improving negative and cognitive symptoms, and these symptom types therefore remain a pressing, unmet medical need [20][22][22,24]. The prevalence, burden and current lack of effective treatments for schizophrenia highlight a need to improve our understanding of the underlying mechanisms and neurobiology of the disorder in order to identify and develop better treatments.
2. Glutamatergic Signalling in the Central Nervous System
Glutamate plays a key role in mediating the homeostatic balance between excitation and inhibition in the brain, cortico-cortical neurotransmission, neuronal development, neurodegeneration, nervous system plasticity and learning and memory [23][37]. Glutamate carries out its actions through its receptors which are divided into two groups: the ionotropic and metabotropic receptors [24][25][38,39]. The ionotropic receptors, namely the NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors, are integral membrane proteins composed of four large subunits (>900 residues) and act as ligand-gated cation channels [26][40]. The metabotropic receptors are G-protein-coupled and activate intracellular biochemical cascades [25][27][39,41].NMDA Receptor Structure and Function
NMDARs are widely distributed and can be found on both neuronal and non-neuronal cells. These receptors play a key role in many physiological processes, including neurodevelopment, synaptogenesis and locomotion, and due to their role as critical mediators of activity-dependent synaptic plasticity, they are especially important for learning, memory formation and other forms of cognition [28][29][30][31][32][33][34][45,46,47,48,49,50,51]. NMDARs are unique in that they require the concomitant binding of glycine and glutamate along with membrane depolarisation for activation and are thus referred to as ‘molecular coincidence detectors’ [35][36][37][52,53,54]. At resting membrane potential, extracellular magnesium (Mg2+) can be found in the ion channel pore, blocking NMDARs in a voltage-dependent manner. Partial depolarisation of the neuron relieves the blockade, opening the channel. The subsequent Ca2+ influx into the neuron triggers a cascade of events that can influence both local, acute functional synaptic plasticity and, via changes in gene expression, sustained neural plasticity [38][55]. The heteromeric composition of NMDARs enables different pharmacological, biophysical and functional properties for the receptor, and these heteromers vary in distribution and expression, both regionally and temporally, throughout development. The different subunits that make up the NMDAR are termed GluN1, GluN2 and GluN3. The GluN1 subunit is encoded by a single gene but has eight splice variants [39][56]. There are four different GluN2 subunits (A–D), encoded by four different genes and two different Glun3 subunits (A and B) [25][40][39,57]. A functional NMDAR is typically composed of two GluN1 subunits and two subunits from among the GluN2A-D and GluN3A-B subunits [28][41][45,58]. The obligatory GluN1 subunit is ubiquitously expressed throughout the brain and over the lifespan [25][42][39,59]. Of the GluN2 subunits, which have a more varied and complex temporal and spatial expression, GluN2A and GluN2B are the predominant subtypes found in the adult human brain, whilst the GluN2C and GluN2D subunits are more highly expressed in the developing brain [28][43][45,60]. The subunit composition of NMDARs influences its functional properties, including agonist affinity, Mg2+ block, decay kinetics and modulation by polyamines [44][45][61,62]. Given the importance of NMDAR subunits in mediating normal brain function, it is not surprising that the dysfunction of these subunits has been linked to various neurological diseases, including schizophrenia. The NMDAR hypofunction model is one of the most commonly adopted and supported models of schizophrenia and is often employed to study the aetiology and pathology of the disorder as well as for the development of novel treatment strategies.3. NMDA Receptor Hypothesis of Schizophrenia
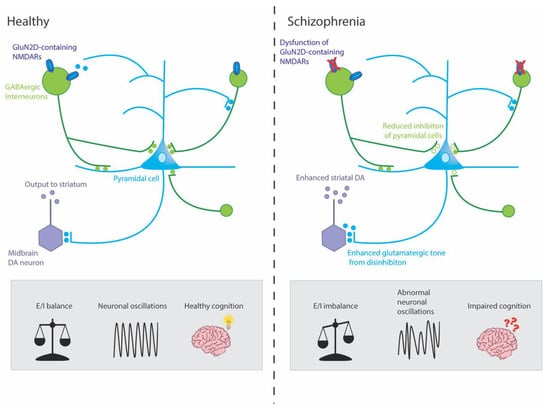
From the late 1950s, phencyclidine (PCP) and ketamine have been reported to induce positive, negative and cognitive phenotypes such as psychosis-like dissociative states and neurocognitive disturbances in healthy individuals like those observed in people with schizophrenia [46][47][48][49][50][51][63,64,65,66,67,68]. Furthermore, these drugs exacerbate symptoms, including psychosis in individuals with schizophrenia [49][52][53][66,69,70]. The NMDAR hypothesis was first proposed soon after, in the late 1970s–1980s, when it was found that PCP and ketamine carry out their actions via NMDAR blockade [47][54][55][56][64,71,72,73]. Although NMDAR hypofunction has been linked to schizophrenia symptoms, the precise underlying mechanisms are still unclear. One hypothesis is that it is primarily the dysfunction of NMDARs on GABAergic interneurons, rather than more widespread NMDAR dysfunction, which contributes to the molecular, physiological and behavioural characteristics of schizophrenia [57][58][35,83]. GABAergic interneurons are stimulated by postsynaptic NMDAR activation and, in turn, synapse onto excitatory glutamatergic pyramidal cells in a negative feedback loop. GABAergic interneurons connect to hundreds of pyramidal cells in this manner, enabling them to coordinate synchronised network activity throughout the brain, including the hippocampus. The activity of these glutamatergic pyramidal cells, in turn, drives downstream striatal dopaminergic neurons (Figure 1) [59][60][84,85].Figure 1. Mechanisms by which dysfunction of GluN2D-containing NMDARs could potentially result in symptoms of schizophrenia. This simplified circuit shows that, in healthy individuals, GluN2D-containing NMDARs stimulate GABAergic interneurons on which they are expressed, which, in turn, mediate inhibition and coordinate the synchronized firing of networks of excitatory pyramidal neurons. These pyramidal neurons, in turn, stimulate dopaminergic (DA) neurons in the midbrain (via the ventral striatum and ventral pallidum, not shown) which project to the associative striatum. In healthy individuals, this circuit is under homeostatic balance, leading to gamma oscillations and healthy cognition. In schizophrenia, hypofunction of the GluN2D-containing NMDARs could lead to GABAergic interneurons increasing excitation of pyramidal neurons by reducing inhibition (disinhibition) onto pyramidal neurons. The resulting aberrant increase in cortical excitation would lead to abnormal neuronal oscillations and impaired cognition. Moreover, this hyperactivity might lead to an overdrive in midbrain DA neurons and enhanced striatal DA, which has been linked to the positive symptoms of schizophrenia.
4. GluN2D Subunit
4.1. GluN2D Receptor Subunit Expression and Distribution
The GluN2D (common names: NMDAR2D, NR2D, GluRε4) subunit is encoded by the GRIN2D gene, which consists of 13 exons, spanning 49.3kB, and is located on chromosome 19q13.1-qter in the human genome [25][39]. The GRIN2D gene has two potential splice isoforms (NR2D-1 and NR2D-2), the longest of which contains 1356 amino acids [25][61][39,113]. In rodents, GluN2D has been extensively characterized; expression levels are first detected between embryonic day (E)15 and 18 during late-embryogenesis, with levels peaking by post-natal day (P)7–10 [62][63][114,115]. The GluN2D mRNA and protein expression levels decrease gradually after the early neonatal phase until late adolescence (P40–50), when they reach their relatively low steady-state expression level [44][62][63][64][61,114,115,116]. During the embryonic and early neonatal phases, the expression of GluN2D is widespread and detected in several regions, including the spinal cord, midbrain nuclei, diencephalon (thalamus, hypothalamus), certain basal ganglia nuclei (substantia nigra and subthalamic nucleus), retina, olfactory bulb, cerebellum, cerebral cortex and hippocampus [43][44][61][62][65][60,61,113,114,117]. The ubiquitous distribution of the GluN2D subunit during the early phases of life makes this subunit particularly interesting amongst the NMDAR subunits as it suggests that GluN2D plays a critical role in modulating circuit connectivity and function during neurodevelopment. This could be of significance when considering a disorder such as schizophrenia that is thought to have its origins in early development [66][67][118,119].
As the rodent ages and GluN2D expression levels reduce, it becomes more localized to distinct cell subtypes [44][62][63][61,114,115]. This is especially apparent in the hippocampus and cortex, where multiple studies have shown that GluN2D is enriched in parvalbumin (PV)-containing GABAergic interneurons in mature rodents, whereas its expression and activity on glutamatergic pyramidal cells decreases [65][68][69][70][71][117,120,121,122,123]. Electrophysiological analyses from the adult mouse medial prefrontal cortex (mPFC) showed that a GluN2C/D positive allosteric modulator, CIQ(+), increased the intrinsic excitability of interneurons and enhanced excitatory postsynaptic currents (EPSCs) from interneurons, whilst not having any effect on the surrounding pyramidal cells [69][121].
In humans, the spatial and temporal expression of GluN2D is thought to be consistent with that reported in rodents; however, it is yet to be well characterised. In human foetal brains, GluN2D mRNA is abundantly expressed and is one of the predominant NMDAR subunits expressed between gestational weeks 8 and 20 [72][126]. In contrast, a study of neurologically normal, adult human post-mortem brains reported only moderate expression of GluN2D mRNA in the prefrontal, parietal and motor cortices, where instead the major subtypes expressed were the GluN2A and GluN2B subunits [73][127]. Theis study also reported that whilst GluN2D expression was low within most neurons in the hippocampus, expression was moderately intense within a small subset of hippocampal neurons, particularly in the hilus, a region containing many interneurons [73][127].
Multiple studies examining the subunit composition of NMDARs in the central nervous system suggest that while the majority of GluN2D subunits are associated with the GluN1 subunit, the GluN2D subunit also forms heteromeric assemblies with GluN2A and/or GluN2B subunits in different brain regions and neuronal subpopulations [74][75][76][129,130,131].