Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Efraím Reyes.
Asymmetric enamine base activation of carbonyl compounds is a well-known and widely used strategy for providing functionalization of organic compounds in an efficient way. The use of solely organic substances, which in most cases are commercially available primary or secondary amines that are easy to obtain, avoids the use of hazardous substances or metal traces, making this type of catalysis a highly convenient methodology from a sustainable point of view.
- dual activation
- aminocatalysis
- asymmetric synthesis
1. Introduction
In 2000, List, Lerner and Barbas III presented the intermolecular cross aldol reaction between acetone and diversely substituted aldehydes promoted by the simple and natural amino acid (S)-proline [1] (Scheme 1a). This was not the first time that this amino acid was employed as the catalyst in an organic transformation [2[2][3],3], but it was presented as a powerful and broad-in-scope catalyst for performing intermolecular cross aldol reactions, also comparable to the best organometallic catalysts for carrying out the same reaction. In this report, based on a previous reaction mechanism of aldolases described by Barbas III [4] and confirmed by computational studies carried out by Houk and List [5], the proposed mechanism included a dual behavior of the catalyst: firstly, it formed an intermediate enamine that increased the nucleophilicity of the α-carbon in acetone, and secondly, it was also involved in the activation of the aldehyde, the second carbonyl unit (Scheme 1b). This catalyst could also provide excellent yields and enantioselectivities using a simple reaction design: stirring the aldehyde and 30 mol% of (S)-proline in a mixture of acetone/DMSO (1:4) for 2–48 h.
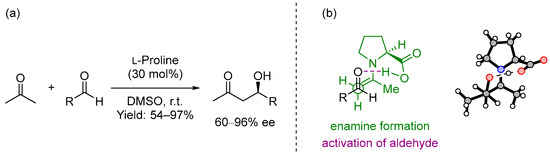
Scheme 1. (a) Proline-catalyzed asymmetric aldol reaction and (b) proposed transition state.
The use of proline as catalyst, together with the well-known MacMillan imidazolidinone introduced the same year [6[6][7],7], represented the starting point in a new research area of great interest: aminocatalysis. This type of catalyst has emerged as a valuable tool for the alleviation of some problems associated with metal catalysis related to trace contamination of those elements. In fact, aminocatalysis [8,9,10,11][8][9][10][11] and other types of organocatalysis [12,13,14,15,16,17,18][12][13][14][15][16][17][18] have been recognized by researchers as one of the most important strategies aligned to green and sustainable catalysis [19]. Aminocatalysis also represents the strategy of choice in many asymmetric transformations, especially when carbonyl compounds are involved, offering good results in terms of yields and enantioselectivities.
In this area, numerous examples of successful reactions have made extensive use of the incorporation of an H-bond donor entity either as an external additive or as a component of a bifunctional aminocatalyst [20,21,22,23,24,25,26][20][21][22][23][24][25][26]. In bifunctional H-bond donor/aminocatalysts, the H-bond donor reagent typically plays a role in the activation of an electrophile that reacts with an enamine-type intermediate. As a result, it becomes a potent stereodirecting component (Scheme 2).
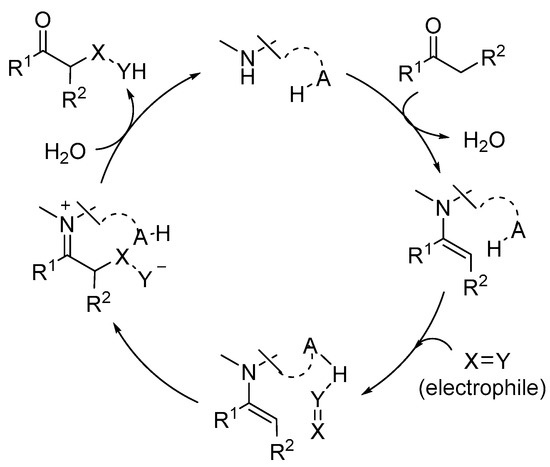
Scheme 2. Illustration of dual activation of reagents in enamine base catalysis.
Alternately, the H-bond donor moiety can also play an important role in events that occur prior to or after the stereogenic center’s installation, either as part of a bifunctional reagent or as an external cocatalyst. This occurs when a Brønsted acidic cocatalyst or stoichiometric additive is added to the reaction mixture to speed up the rate of catalyst turnover during the hydrolysis step or to make it easier to activate the carbonyl group of the aldehyde or ketone substrate during the condensation with the aminocatalyst in the activation stage. This science is beyond the scope of this paper and will not be examined exhaustively.
2. Dual H-Bonding and Enamine Activation
An excellent illustration of the involvement of an H-bond donor site in the structure of the aminocatalyst during the aldol reaction between the intermediate enamine and the external aldehyde reagent is the pioneering proline-catalyzed aldol reaction (Scheme 3). As a result, outstanding levels of both diastereo- and enantioselectivity are achieved as the reaction progresses through a cyclic chair-like transition state with reduced conformational mobility. This generated H-bond during the C-C bond forming step has been used as a general strategy during experimental and computational studies [5,27,28,29,30,31,32,33,34][5][27][28][29][30][31][32][33][34]. In fact, the conformational restricted transition state does not just record the extremely high facial selectivity; this situation restricts the E diastereoisomer formed in the transient enamine species, as the presence of serious dynamic and thermodynamic restrictions in the less favored Z diastereoisomer (see Scheme 2).
Scheme 3. General proline-catalyzed cross-aldol reaction.
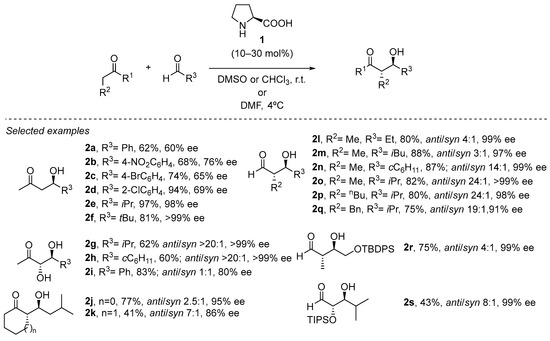
This transformation is rather wide in scope with respect to the possibility of using different aldehydes in combination with acetone [35[35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56],36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56], α-hydroxyketones [57,58][57][58] or cyclic ketones [59] as the pronucleophile source, including the possibility of performing the reaction at multigram or even kilogram scale. Moreover, the reaction can also be carried out in open air and without the need for degassed or dry solvent, which is an important benefit in terms of operational simplicity. In addition, the cross-aldol reaction between two different aldehydes can also be carried out under slightly modified conditions [60], also enabling the use of polyhydroxylated aldehydes as either pronucleophiles or electrophiles [61], which also opens the possibility for the stereoselective synthesis of sugars through this methodology [62].
The same stereochemical model can be applied to structurally related electrophiles, thus expanding the portfolio of organic transformations in which this approach can be applied [63,64][63][64]. In particular, proline has been demonstrated to be an outstanding catalyst for performing the Mannich reaction (Scheme 4) [65,66,67,68,69][65][66][67][68][69]. In this case, the availability of a single electron pair at the nitrogen atom available to engage in the H-bonding interaction with the carboxylate group of the proline leads to the formation of the syn diastereoisomer as a consequence of the (E) geometry of the azomethine moiety, in contrast to the anti diastereoselection previously observed in the parent aldol reaction (see Scheme 4). Also, the possibility of performing the challenging cross-Mannich reaction using acetaldehyde as the pronucleophile should be highlighted, providing acceptable yields of the corresponding α-unsubstituted β-aminoaldehydes and minimizing to a great extent the presence of byproducts arising from competitive polymerization through self-aldol condensation [70].
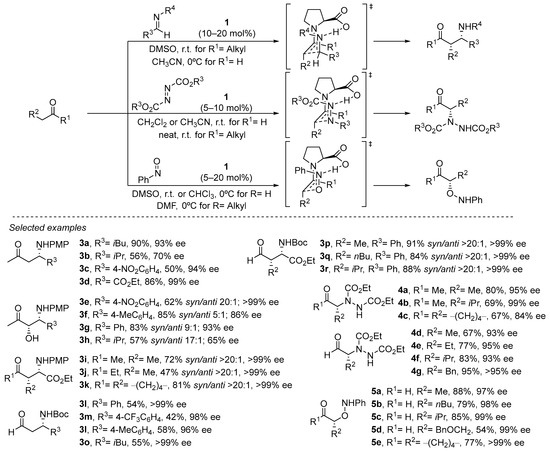
Scheme 4. Proline-catalyzed α-functionalization reactions.
Other electronically similar electrophiles such as azodicarboxylates or nitrosoarenes also perform excellently in the α-amination [71,72,73,74,75,76][71][72][73][74][75][76] and the α-hydroxylation [77,78,79,80,81,82,83][77][78][79][80][81][82][83] (or nitroso-aldol reaction) of aldehydes and ketones under proline catalysis, providing an efficient and direct entry to enantioenriched α-aminocarbonyl or α-hydroxycarbonyl compounds or related derivatives (See Scheme 4). In the latter case, the participation of such an H-bonding interaction with the electrophile also conditions the chemoselectivity of the reaction, observing that Jørgensen–Hayashi catalysts that do not contain any H-bond donor motif also efficiently catalyze the reaction between aldehydes and nitrosobenzene but lead to the formation of the opposite N-addition isomer, thus resulting in the α-hydroxyamination of the starting material [84].
There are still a few situations in which the reaction is limited due to either poor enantiocontrol or low conversion, despite the fact that proline performs exceptionally well in many of these reactions and has a rather broad substrate scope. This has been credited by and large to the unfortunate dissolvability of proline in the normal natural solvents utilized for the response. A possible solution has been found through the incorporation of achiral H-bond donor additives able to engage in H-bonding interactions with the carboxylate moiety that can increase the solubility of the catalyst and also contribute to the activation of the electrophile through the H-bonding interactions. This is the case of the combined use of l-proline and chiral BINOL 6, which, used in the correct matched combination and under optimized ratio, provided a remarkable improvement in the aldol reaction between acetone and several aromatic aldehydes compared to the parent reaction without any additive (Scheme 5) [38]. Further research on this behavior has led to improved systems that combine the use of l-proline with an external achiral thiourea cocatalyst [47,85,86,87][47][85][86][87] as H-bond donor.
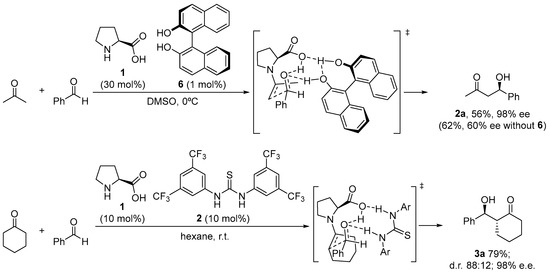
Scheme 5. Proline/H-bond additive catalyzed asymmetric aldol reaction.
Obviously, proline is not a universal catalyst for this type of transformation, and a wide variety of structurally related catalysts based on the proline scaffold have been studied and employed with different degrees of success in aldol [88], Mannich [89,90[89][90][91],91], α-amination [92] or α-aminoxylation [93] involving the enamine activation of aldehydes and ketones. The logical evolution of the initial system relied on the modification of the hydrogen-bond donor moiety, also tuning its acidity and availability or incorporating multiple additional H-bond donor units within its structure [94,95][94][95]. Some selected examples that provide an overall view of the different directions taken in this area are displayed in Figure 1. For instance, simply changing from the carboxylate moiety on proline to the corresponding N-arylamide 7 [96,97,98][96][97][98] or N-sulfonylamide 8 [99,100,101,102,103,104,105][99][100][101][102][103][104][105] leads to competent catalysts in such transformations. The same applies to the substitution of the carboxylate with other related motifs, as in the case of prolinamide 9 [106,107,108,109][106][107][108][109] or pyrrolidine-tetrazole catalyst 10 [110,111,112,113,114][110][111][112][113][114] or the possibility of using modified versions of trans-4-hydroxyproline as in the case of compound 11 [115,116][115][116]. As alternatives, several authors have also surveyed the incorporation of substituents with additional stereogenic elements like α-amino acids [117,118[117][118][119][120],119,120], sugar moieties [121] or other, more complex alkaloids [122,123,124][122][123][124] (see, for example, catalysts 12, 13 and 14). Another strategy has also relied on incorporating functionalities with additional H-bond donor motifs that lead to the formation of a transition state in which both reagents, the enamine intermediate and the electrophile are connected through a network of H-bonding interactions that turn into reduced conformational mobility. Some examples include the use of aminoalcohol-derived prolinamide 15 [96,125,126][96][125][126] or diamine-based catalysts such as 16 [127,128][127][128] or 17 [129,130,131][129][130][131] that also incorporate additional stereogenic elements on the N-substituent or even with a terminal thiourea moiety that provides enhanced H-bond donor ability [132,133,134,135,136][132][133][134][135][136] (see catalyst 18 for a representative example). Alternatively, dipeptide-type catalysts [137,138,139][137][138][139] with a terminal secondary amide moiety (like in catalysts 19 [140] or 20 [141]) or more complex polypeptidic scaffolds (see catalysts 21 [142] and 22 [143] as examples), which are based on either natural or unnatural [144] amino acid scaffolds, have also proved to be useful in this type of reaction (23) [145].
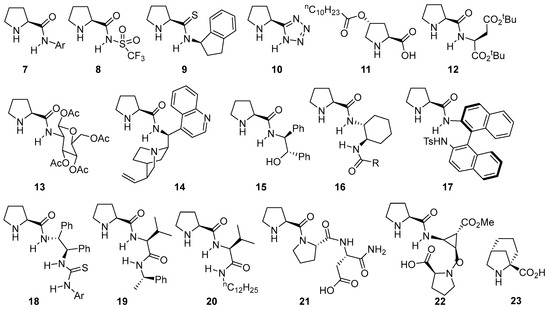
Figure 1. Selected examples of bifunctional proline-based aminocatalysts incorporating H-bond donor moieties employed in aldol, Mannich and related reactions.
Interestingly, moving the carboxylate group in the pyrrolidine core from the 2-position to the 3-position leads to a huge difference in how the electrophile and the enamine intermediate organize in the transition state. For instance, the use of pyrrolidine-3-carboxylate 24 in the Mannich reaction leads to a complete change in the simple diastereoselection of the reaction, moving from the syn-selectivity reported for the proline-catalyzed reaction (see Scheme 4) to provide the anti Mannich adducts in excellent yield and stereocontrol (Scheme 6) [146]. This behavior was explained through the formation of an H-bonded intermediate in which the chair-like TS operating in the l-proline-catalyzed reaction—in which the H-bonded imine approaches the reactive (E)-s-trans enamine conformer—was no longer operating, thus moving to a situation in which—while the carboxylate still directs the incoming electrophile from the same face of the (E)-enamine intermediate—this enamine has changed its reactive conformation to s-cis, thus exposing the opposite diastereotopic face. A similar behavior has also been described for related catalyst systems based on trans-4-hydroxyproline [147,148][147][148] or trans-3-aminoproline [149,150][149][150].
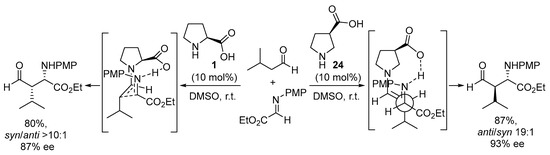
Scheme 6. The proline- vs. 3-pyrrolidinecarboxylate-catalyzed enantioselective Mannich reactions.
A similar effect in which internal H-bonding changes the reactive conformation of the enamine intermediate in order to provide opposite simple diastereoselection to those observed in the archetypical l-proline-catalyzed aldol or Mannich reactions has been explored by Maruoka and coworkers with bifunctional catalyst 25a based on the binaphthyl core (Scheme 7) [151,152,153,154,155,156][151][152][153][154][155][156]. In this case, an acidic sulfonamide substituent at the 3-position of the binaphthyl core plays the role of the stereodirecting element as an H-bond donor motif.
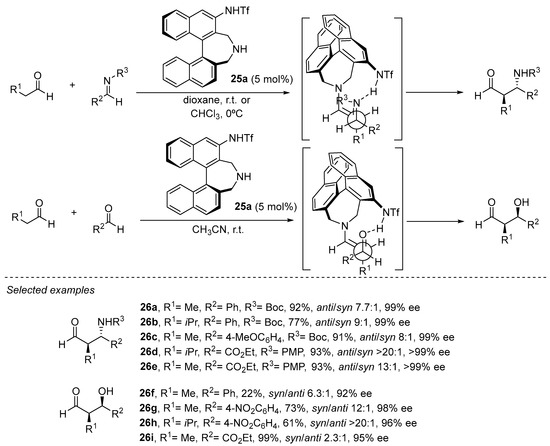
Scheme 7. Binaphthyl-based chiral catalyst 24a in enantioselective aldol and Mannich reactions.
Primary amines have been demonstrated to perform well in aldol, Mannich, α-amination and α-aminoxylation reactions [133,157,158][133][157][158]. The enamine formed after condensation with a primary amine has a reduced degree of steric congestion around the enamine moiety and, therefore, enables the activation of more sterically demanding substrates such as, for example, acyclic ketones or α,α-disubstituted aldehydes. In addition, the NH moiety of the secondary enamine is able to engage in H-bonding interactions with the incoming electrophile or with additional Lewis basic sites of the catalysts, facilitating the formation of a geometrically defined intermediate and, therefore, a higher degree of stereocontrol. In fact, the smallest natural chiral α-amino acid, L-alanine (27), has demonstrated its proficiency in catalyzing the aldol reaction (Scheme 8) [159], and other reports afterwards demonstrated that several other proteinogenic α-amino acids were also good catalysts for aldol and related reactions [160].
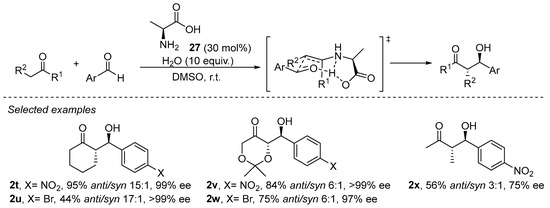
Scheme 8. The L-alanine-catalyzed enantioselective aldol reaction.
There are also several other bifunctional catalysts involving primary amines that incorporate additional H-bond donor entities reported for these transformations (see Figure 2 for several representative examples), starting with simple modifications of the a-amino acid core like, for example 28 [161]), chiral diamine-based compounds like 29 [162] or 30 [163] and also dipeptides, tripeptides or even longer peptide compounds (illustrative cases: 31 [164], 32 [165,166][165][166] and 33 [167]).
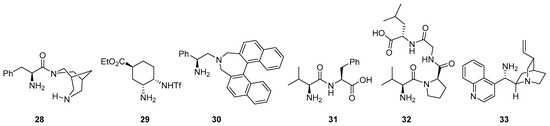
Figure 2. Selected examples of bifunctional primary amine/H-bond donor catalysts employed in aldol, Mannich and related reactions.
The use of α,β-unsaturated carbonyl compounds as electrophiles in the Michael reaction with aldehydes and ketones under enamine catalysis deserves special attention [168]. The fact that the Lewis basic site at the Michael acceptor that has to interact with the H-bond donor motif of the catalyst is placed at a longer distance in comparison with the previously discussed electrophiles entails that most of the bifunctional hydrogen bond donor/aminocatalysts that perform well in the aldol, Mannich, α-hydrazination or α-aminoxylation tend to provide poor results in the Michael reaction. For example, the l-proline-catalyzed Michael reaction between cyclohexanone and β-nitrostyrene proceeds and provides the corresponding Michael adduct in excellent yield and diastereoselectivity but with poor enantiocontrol [169], and the same applies to tetrazole analogue 10 [170], but increasing the distance between the secondary amine moiety and the H-bond donor site, as in homoproline/tetrazole catalyst 34, led to a significant improvement in the enantioselectivity (Scheme 9) [171]. In this sense, the well-known ability of thioureas to establish persistent interactions through double H-bonding events with the nitro group also has been applied to the design of efficient bifunctional pyrrolidine/thiourea catalysts such as 35 [172[172][173],173], which also performs well in the Michael reaction using nitroalkenes as electrophiles [174,175,176,177,178][174][175][176][177][178].
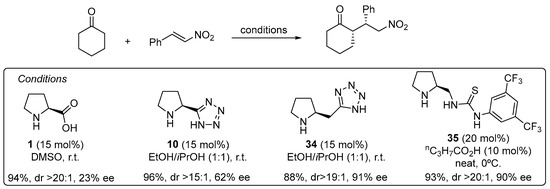
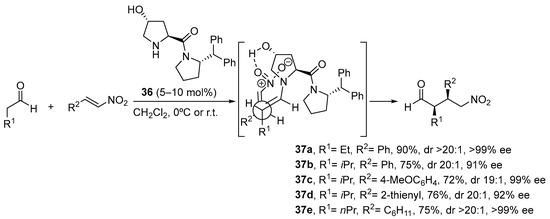
Scheme 9. The Michael reaction between cyclohexanone and β-nitrostyrene catalyzed by proline and tetrazolylpyrrolidine vs. functionalized pyrrolidines 34 and 35. As an alternative, trans-4-hydoxy-l-prolinamide 36 has also been described to promote very efficiently the enantioselective Michael reaction between aldehydes and nitroalkenes (Scheme 10) [179]. In this case, the facial selectivity is reversed with respect to the reactions catalyzed by l-proline or the related derivatives shown in the previous scheme, which results from the internal activation of the nitroalkene by the 4-OH moiety of the enamine intermediate. The bulky amide substituents also contribute to enhance the enantioselectivity of the reaction through favoring the selective formation of the (E)-s-trans conformer in which steric interactions are minimized.
Scheme 10. The Michael reaction between aldehydes and nitroalkenes catalyzed by trans-hydroxyprolinamide 36.
On the other hand, tripeptide 38a has been demonstrated to be, up to date, the best performing catalyst in this transformation, being able to catalyze the transformation in a remarkably low 1 mol% catalyst loading (Scheme 11) [180]. The reaction can be carried out on a remarkably wide set of aldehyde donors and nitroalkene Michael acceptors, including the highly challenging nitroethylene [181] and also α,β- and β,β-disubstituted nitroalkenes [182,183][182][183]. In addition, the reaction proceeds under almost equimolar amounts of both Michael donor and acceptor, in deep contrast with most reported methodologies that typically required a large excess of the nitroalkene. Several key elements are required for the high performance of this catalyst. On one hand, the presence of the secondary pyrrolidine from the initial d-proline residue is crucial for both activity and enantioselectivity, and the configuration of the other two L-amino acid residues has also been recognized as the matched combination that provides the highest enantiocontrol [184]. On the other hand, the terminal carboxylic acid moiety of the final glutamic acid residue was also identified to be key for both activity and enantioselectivity [185]. Interestingly, reducing the size of this final carboxylate-containing side chain (from glutamic acid to aspartic acid) also led to a slight decrease in yield and enantioselectivity. A series of in-depth mechanistic studies [186,187,188,189,190,191,192][186][187][188][189][190][191][192] indicate that this carboxylic acid moiety is engaged in H-bonding interactions with the nitroalkene during the Michael addition between the intermediate enamine and the nitroalkene, but this acidic moiety is also crucial for promoting the fast protonation of the nitronate intermediate formed after the conjugate addition step. This favors catalyst turnover and leaves the C-C bond-forming step as the stereodefining event in the catalytic cycle [193]. Further studies in this area by other authors have shown that other structurally related tripeptides [194,195,196][194][195][196] or smaller dipeptides [197,198][197][198] also can catalyze this reaction with success.
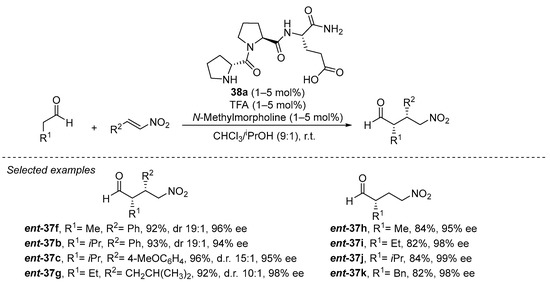
Scheme 11. The D-Pro-Pro-Glu-NH2 (38a)-catalyzed Michael reaction between aldehydes and nitroalkenes.
As an interesting variant, catalyst 38b, in which steric bulk at C4 of the active proline residue has been increased through the introduction of two methyl substituents, is an outstanding catalyst for the same Michael reaction, providing the opposite anti diastereoisomer with excellent yield and enantioselectivity (Scheme 12) [199]. The basis for this change in diastereoselectivity relies on the change in reactive conformation of the enamine intermediate, which in this case adopts a s-cis conformation in order to avoid steric clash between the alkene moiety and the two methyl substituents.
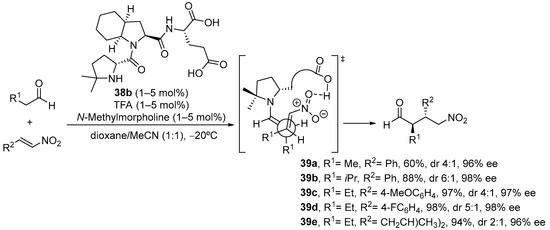
Scheme 12. The tripeptide 38b-catalyzed Michael reaction between aldehydes and nitroalkenes providing anti diastereoisomers.
The use of acyclic ketones as Michael donors typically makes use of primary amines as catalysts in order to favor the enamine intermediate with a lower degree of steric congestion compared to the situation when secondary amines are used. In this field, bifunctional primary amine/thioureas have gained a prominent position among the different systems reported to promote the Michael reaction between ketones and nitroalkenes. For example, 1,2-diphenylethylenediamine-based thiourea 40a has demonstrated its proficiency in this transformation with both acetone and other substituted acyclic ketones [200,201][200][201], in the latter case leading to the diastereoselective formation of the anti γ-nitro ketone diastereoisomer with high enantioselectivity (Scheme 13). This is explained in terms of the preferential participation of the Z enamine intermediate that avoids destabilizing steric interactions between the two alkyl substituents across the C=C bond. Moreover, in this case the reaction was also found to be completely regioselective, providing α-branched adduct 41e and without observing the formation of the potential regioisomer arising through the competitive formation of an unsubstituted enamine intermediate upon condensation of the substrate with the catalyst. Further progress in this field has evolved into a wide variety of structurally related primary amine/thiourea catalysts that also perform well in this transformation [144].
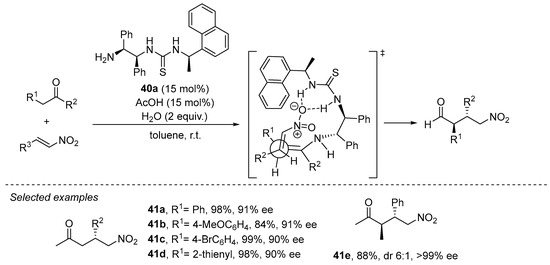
Scheme 13. The Michael reaction between acyclic ketones and nitroalkenes catalyzed by primary amine/thiourea 40a.
The same situation shows up when α,α-disubstituted aldehydes are to be used as Michael donors [202], in which the problems associated with the high degree of steric congestion around the nucleophilic carbon of the enamine are circumvented through the use of a primary amine catalyst instead of the archetypical pyrrolidine-based secondary amines. A particularly efficient approach comprises the use of bifunctional trans-cyclohexanediamine-derived thiourea 42a (Scheme 14) [203]. As can be seen in this scheme, this reaction performs excellently for a variety of situations, including challenging β-alkyl substituted nitroalkenes and also functionalized aldehydes, providing generally excellent yields and stereocontrol. It should also be pointed out that this catalyst is also particularly efficient in the Michael reaction between acyclic ketones and nitroalkenes [204].
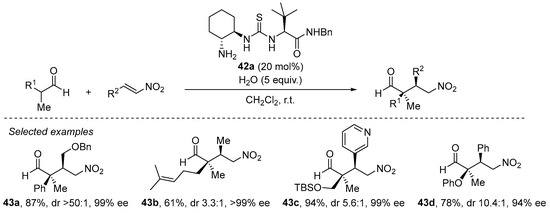
Scheme 14. The Michael reaction between α,α-disubstituted aldehydes and nitroalkenes catalyzed by primary amine/thiourea 42a.
References
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396.
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621.
- Eder, U.; Sauer, G.; Wiecert, R. New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Angew. Chem. Int. Ed. 1971, 10, 496–497.
- Wagner, J.; Lerner, R.A.; Barbas, C.F., III. Efficient Aldolase Catalytic Antibodies That Use the Enamine Mechanism of Natural Enzymes. Science 1995, 270, 1797–1800.
- Bahmanyar, S.; Houk, K.N.; Martin, H.J.; List, B. Quantum Mechanical Predictions of the Stereoselectivities of Proline-Catalyzed Asymmetric Intermolecular Aldol Reactions. J. Am. Chem. Soc. 2003, 125, 2475–2479.
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels-Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244.
- Jen, W.S.; Wiener, J.J.M.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Enantioselective Organocatalytic 1,3-Dipolar Cycloaddition. J. Am. Chem. Soc. 2000, 122, 9874–9875.
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569.
- Sulzer-Mossé, S.; Alexakis, A. Chiral amines as organocatalysts for asymmetric conjugate addition to nitroolefins and vinyl sulfonesviaenamine activation. Chem. Commun. 2007, 30, 3123–3135.
- Kano, T.; Maruoka, K. Design of chiral bifunctional secondary amine catalysts for asymmetric enamine catalysis. Chem. Commun. 2008, 43, 5465–5473.
- Melchiorre, P. Cinchona-based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770.
- Xiang, S.-H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786.
- List, B.; Maruoka, K. (Eds.) Science of Synthesis, Asymmetric Organocatalysis; Georg Thieme Verlag: Stuttgart, Germany, 2012.
- Pellissier, H. (Ed.) Recent Developments in Asymmetric Organocatalysis; RSC Publishing: Cambridge, UK, 2011.
- Jacobsen, E.N.; MacMillan, D.W.C. Organocatalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 20618–20619.
- Bertelsen, S.; Jørgensen, K.A. Organocatalysis-after the gold rush. Chem. Soc. Rev. 2009, 38, 2178–2189.
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308.
- Holland, M.C.; Gilmour, R. Deconstructing Covalent Organocatalysis. Angew. Chem. Int. Ed. 2015, 54, 3862–3871.
- Sihtmae, M.; Silm, M.; Kriis, K.; Kahru, A.; Kanger, T. Aminocatalysts are More Environmentally Friendly than Hydrogen-Bonding Catalysts. ChemSusChem 2022, 15, e202201045.
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from Molecular Recognition to Bifunctional Organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899.
- Matos Paz, B.; Jiang, H.; Jørgensen, K.A. Aminocatalysis: Beyond Steric Shielding and Hydrogen-Bonding. Chem. Eur. J. 2015, 21, 1846–1853.
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743.
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional Amine-Squaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions. Adv. Synth. Catal. 2015, 357, 253–281.
- Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Additive Effects on Asymmetric Catalysis. Chem. Rev. 2016, 116, 4006–4123.
- Anebouselvy, K.; Shruthi, K.S.; Ramachary, D.B. Asymmetric Supramolecular Organocatalysis: A Complementary Upgrade to Organocatalysis. Eur. J. Org. Chem. 2017, 2017, 5460–5483.
- Meeuwissen, J.; Reek, J.N.H. Supramolecular catalysis beyond enzyme mimics. Nature Chem. 2010, 2, 615–621.
- Allemann, C.; Gordillo, R.; Clemente, F.R.; Cheong, P.H.-Y.; Houk, K.N. Theory of Asymmetric Organocatalysis of Aldol and Related Reactions: Rationalizations and Predictions. Acc. Chem. Res. 2004, 37, 558–569.
- Hoang, L.; Bahmanyar, S.; Houk, K.N.; List, B. Kinetic and Stereochemical Evidence for the Involvement of Only One Proline Molecule in the Transition States of Proline-Catalyzed Intra- and Intermolecular Aldol Reactions. J. Am. Chem. Soc. 2003, 125, 16–17.
- List, B.; Hoang, L.; Martin, H.J. New mechanistic studies on the proline-catalyzed aldol reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 5839–5842.
- Orlandi, M.; Ceotto, M.; Benaglia, M. Kinetics versus thermodynamics in the proline catalyzed aldol reaction. Chem. Sci. 2016, 7, 5421–5427.
- Sharma, A.K.; Sunoj, R.B. Enamine versus Oxazolidinone: What Controls Stereoselectivity in Proline-Catalyzed Asymmetric Aldol Reactions? Angew. Chem. Int. Ed. 2010, 49, 6373–6377.
- Schmid, M.B.; Zeitler, K.; Gschwind, R.M. The Elusive Enamine Intermediate in Proline-Catalyzed Aldol Reactions: NMR Detection, Formation Pathway, and Stabilization Trends. Angew. Chem. Int. Ed. 2010, 49, 4997–5003.
- Clemente, F.R.; Houk, K.N. Computational Evidence for the Enamine Mechanism of Intramolecular Aldol Reactions Catalyzed by Proline. Angew. Chem. Int. Ed. 2004, 43, 5766–5768.
- Haindl, M.H.; Hioe, J.; Gschwind, R.M. The Proline Enamine Formation Pathway Revisited in Dimethyl Sulfoxide: Rate Constants Determined via NMR. J. Am. Chem. Soc. 2015, 137, 12835–12842.
- Martínez, A.; Zumbansen, K.; Döhring, A.; van Gemmeren, M.; List, B. Improved Conditions for the Proline-Catalyzed Aldol Reaction of Acetone with Aliphatic Aldehydes. Synlett 2014, 25, 932–934.
- Martínez-Castañeda, A.; Poladura, B.; Rodríguez-Solla, H.; Concellón, C.; del Amo, V. Direct Aldol Reactions Catalyzed by a Heterogeneous Guanidinium Salt/Proline System under Solvent-Free Conditions. Org. Lett. 2011, 13, 3032–3035.
- Doyagüez, E.G.; Calderón, F.; Sánchez, F.; Fernández-Mayoralas, A. Asymmetric Aldol Reaction Catalyzed by a Heterogenized Proline on a Mesoporous Support. The Role of the Nature of Solvents. J. Org. Chem. 2007, 72, 9353–9356.
- Zhou, Y.; Shan, Z. Chiral Diols: A New Class of Additives for Direct Aldol Reaction Catalyzed by l-Proline. J. Org. Chem. 2006, 71, 9510–9512.
- Pan, Q.; Zou, B.; Wang, Y.; Ma, D. Diastereoselective Aldol Reaction of N,N-Dibenzyl-α-amino Aldehydes with Ketones Catalyzed by Proline. Org. Lett. 2004, 6, 1009–1012.
- Itoh, T.; Yokoya, M.; Miyauchi, K.; Nagata, K.; Ohsawa, A. Proline-Catalyzed Asymmetric Addition Reaction of 9-Tosyl-3,4-dihydro-β-carboline with Ketones. Org. Lett. 2003, 5, 4301–4304.
- Cordova, A.; Notz, W.; Barbas , C.F., III. Proline-Catalyzed One-Step Asymmetric Synthesis of 5-Hydroxy-(2E)-hexenal from Acetaldehyde. J. Org. Chem. 2002, 67, 301–303.
- Villano, R.; Rosaria Acocella, M.; Scettri, A. Influence of a remote sulfinyl group on l-proline-catalyzed direct asymmetric aldol addition of acetone. Tetrahedron 2016, 72, 5414–5419.
- Llanes, P.; Sayalero, S.; Rodríguez-Escrich, C.; Pericàs, M.A. Asymmetric cross- and self-aldol reactions of aldehydes in water with a polystyrene-supported triazolylproline organocatalyst. Green Chem. 2016, 18, 3507–3512.
- Demir, A.S.; Basceken, S. Study of asymmetric aldol and Mannich reactions catalyzed by proline–thiourea host–guest complexes in nonpolar solvents. Tetrahedron Asymmetry 2013, 24, 515–525.
- Montroni, E.; Sanap, S.P.; Lombardo, M.; Quintavalla, A.; Trombini, C.; Dhavale, D.D. A New Robust and Efficient Ion-Tagged Proline Catalyst Carrying an Amide Spacer for the Asymmetric Aldol Reaction. Adv. Synth. Catal. 2011, 353, 3234–3240.
- Karmakar, A.; Maji, T.; Wittmann, S.; Reiser, O. l-Proline/CoCl2-Catalyzed Highly Diastereo- and Enantioselective Direct Aldol Reactions. Chem. Eur. J. 2011, 17, 11024–11029.
- El-Hamdouni, N.; Companyó, X.; Rios, R.; Moyano, A. Substrate-Dependent Nonlinear Effects in Proline–Thiourea-Catalyzed Aldol Reactions: Unraveling the Role of the Thiourea Co-Catalyst. Chem. Eur. J. 2010, 16, 1142–1148.
- Shah, J.; Blumenthal, H.; Yacob, Z.; Liebscher, J. Proline-Catalyzed Asymmetric Aldol Reaction in Guanidine- Derived Ionic Liquids. Adv. Synth. Catal. 2008, 350, 1267–1270.
- Pihko, P.M.; Laurikainen, K.M.; Usano, A.; Nyberg, A.I.; Kaavi, J.A. Effect of additives on the proline-catalyzed ketone–aldehyde aldol reactions. Tetrahedron 2006, 62, 317–328.
- Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Sumiya, T.; Urushima, T.; Shoji, M.; Hashizume, D.; Koshino, H. A Highly Active 4-Siloxyproline Catalyst for Asymmetric Synthesis. Adv. Synth. Catal. 2004, 346, 1435–1439.
- Pihko, P.M.; Erkkilä, A. Enantioselective synthesis of prelactone B using a proline-catalyzed crossed-aldol reaction. Tetrahedron Lett. 2003, 44, 7607–7609.
- Kotrusz, P.; Kmentová, I.; Gotov, B.; Toma, S.; Solčániová, E. Proline-catalysed asymmetric aldol reaction in the room temperature ionic liquid PF6. Chem. Commun. 2002, 8, 2510–2511.
- Pidathala, C.; Hoang, L.; Vignola, N.; List, B. Direct Catalytic Asymmetric Enolexo Aldolizations. Angew. Chem. Int. Ed. 2003, 42, 2785–2788.
- Chandler, C.L.; List, B. Catalytic, Asymmetric Transannular Aldolizations: Total Synthesis of (+)-Hirsutene. J. Am. Chem. Soc. 2008, 130, 6737–6739.
- Cordova, A.; Notz, W.; Barbas, C.F., III. Direct organocatalytic aldol reactions in buffered aqueous media. Chem. Commun. 2002, 24, 3024–3025.
- Hayashi, Y.; Aratake, S.; Itoh, T.; Okano, T.; Sumiya, T.; Shoji, M. Dry and wet prolines for asymmetric organic solvent-free aldehyde–aldehyde and aldehyde–ketone aldol reactions. Chem. Commun. 2007, 9, 957–959.
- Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C.F., III. Amino Acid Catalyzed Direct Asymmetric Aldol Reactions: A Bioorganic Approach to Catalytic Asymmetric Carbon−Carbon Bond-Forming Reactions. J. Am. Chem. Soc. 2001, 123, 5260–5267.
- Notz, W.; List, B. Catalytic Asymmetric Synthesis of anti-1,2-Diols. J. Am. Chem. Soc. 2000, 122, 7386–7387.
- List, B.; Pojarliev, P.; Castello, C. Proline-Catalyzed Asymmetric Aldol Reactions between Ketones and α-Unsubstituted Aldehydes. Org. Lett. 2001, 3, 573–575.
- Northrup, A.B.; MacMillan, D.W.C. The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes. J. Am. Chem. Soc. 2002, 124, 6798–6799.
- Northrup, A.B.; Mangion, F.H.; MacMillan, D.W.C. Enantioselective Organocatalytic Direct Aldol Reactions of α-Oxyaldehydes: Step One in a Two-Step Synthesis of Carbohydrates. Angew. Chem. Int. Ed. 2004, 43, 2152–2154.
- Northrup, A.B.; MacMillan, D.W.C. Two-step synthesis of carbohydrates by selective aldol reactions. Science 2004, 305, 1752–1755.
- Notz, W.; Tanaka, F.; Barbas, C.F., III. Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions. Acc. Chem. Res. 2004, 37, 580–591.
- List, B. Enamine Catalysis Is a Powerful Strategy for the Catalytic Generation and Use of Carbanion Equivalents. Acc. Chem. Res. 2004, 37, 548–557.
- Cordova, A.; Notz, W.; Zhong, G.; Betancort, J.; Barbas, C.F., III. A Highly Enantioselective Amino Acid-Catalyzed Route to Functionalized α-Amino Acids. J. Am. Chem. Soc. 2002, 124, 1842–1843.
- List, B.; Pojarliev, P.; Biller, W.T.; Martin, H.J. The Proline-Catalyzed Direct Asymmetric Three-Component Mannich Reaction: Scope, Optimization, and Application to the Highly Enantioselective Synthesis of 1,2-Amino Alcohols. J. Am. Chem. Soc. 2002, 124, 827–833.
- List, B. The Direct Catalytic Asymmetric Three-Component Mannich Reaction. J. Am. Chem. Soc. 2000, 122, 9336–9337.
- Yang, J.W.; Stadler, M.; List, B. Proline-Catalyzed Mannich Reaction of Aldehydes with N-Boc-Imines. Angew. Chem. Int. Ed. 2007, 46, 609–611.
- Hayashi, Y.; Urushima, T.; Tsuboi, W.; Shoji, M. l-Proline-catalyzed enantioselective one-pot cross-Mannich reaction of aldehydes. Nat. Protoc. 2007, 2, 113–118.
- Yang, J.; Chandler, C.; Stadler, M.; Kempen, D.; List, B. Proline-catalysed Mannich reactions of acetaldehyde. Nature 2008, 452, 453–455.
- Bøgevig, A.; Juhl, K.; Kumaragurubaran, N.; Zhuang, W.; Jørgensen, K.A. Direct Organo-Catalytic Asymmetric α-Amination of Aldehydes—A Simple Approach to Optically Active α-Amino Aldehydes, α-Amino Alcohols, and α-Amino Acids. Angew. Chem. Int. Ed. 2002, 41, 1790–1793.
- List, B. Direct Catalytic Asymmetric α-Amination of Aldehydes. J. Am. Chem. Soc. 2002, 124, 5656–5657.
- Kumaragurubaran, N.; Juhl, K.; Zhuang, W.; Bøgevig, A.; Jørgensen, K.A. Direct l-Proline-Catalyzed Asymmetric α-Amination of Ketones. J. Am. Chem. Soc. 2002, 124, 6254–6255.
- Juhl, K.; Jørgensen, K.A. Catalytic Asymmetric Direct α-Amination Reactions of 2-Keto Esters: A Simple Synthetic Approach to Optically Active syn-β-Amino-α-hydroxy Esters. J. Am. Chem Soc. 2002, 124, 2420–2421.
- Ashley, M.A.; Hirschi, J.S.; Izzo, J.A.; Vetticatt, M.J. Isotope Effects Reveal the Mechanism of Enamine Formation in l-Proline-Catalyzed α-Amination of Aldehydes. J. Am. Chem. Soc. 2016, 138, 1756–1759.
- Kanzian, T.; Lakhdar, S.; Mayr, H. Kinetic Evidence for the Formation of Oxazolidinones in the Stereogenic Step of Proline-Catalyzed Reactions. Angew. Chem. Int. Ed. 2010, 49, 9526–9529.
- Brown, S.P.; Brochu, M.P.; Sinz, C.J.; MacMillan, D.W.C. The Direct and Enantioselective Organocatalytic α-Oxidation of Aldehydes. J. Am. Chem. Soc. 2003, 125, 10808–10809.
- Zhong, G. A Facile and Rapid Route to Highly Enantiopure 1,2-Diols by Novel Catalytic Asymmetric α-Aminoxylation of Aldehydes. Angew. Chem. Int. Ed. 2003, 42, 4247–4250.
- Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Shoji, M. Direct Proline-Catalyzed Asymmetric α-Aminoxylation of Ketones. Angew. Chem. Int. Ed. 2004, 43, 1112–1115.
- Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Hibino, K.; Shoji, M. Direct Proline-Catalyzed Asymmetric α-Aminoxylation of Aldehydes and Ketones. J. Org. Chem. 2004, 69, 5966–5973.
- Bogevig, A.; Sunden, H.; Cordova, A. Direct Catalytic Enantioselective α-Aminoxylation of Ketones: A Stereoselective Synthesis of α-Hydroxy and α,α′-Dihydroxy Ketones. Angew. Chem. Int. Ed. 2004, 43, 1129–1132.
- Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Shoji, M. Direct proline catalyzed asymmetric α-aminooxylation of aldehydes. Tetrahedron Lett. 2003, 44, 8293–8296.
- Kumar, P.; Dwivedi, N. Proline Catalyzed α-Aminoxylation Reaction in the Synthesis of Biologically Active Compounds. Acc. Chem. Res. 2013, 46, 289–299.
- Palomo, C.; Vera, S.; Velilla, I.; Mielgo, A.; Gomez-Bengoa, E. Regio- and Enantioselective Direct Oxyamination Reaction of Aldehydes Catalyzed by α,α-Diphenylprolinol Trimethylsilyl Ether. Angew. Chem. Int. Ed. 2007, 46, 8054–8056.
- Companyó, X.; Valero, G.; Crovetto, L.; Moyano, A.; Rios, R. Highly Enantio- and Diastereoselective Organocatalytic Desymmetrization of Prochiral Cyclohexanones by Simple Direct Aldol Reaction Catalyzed by Proline. Chem. Eur. J. 2009, 15, 6564–6568.
- Reis, O.; Eymur, S.; Reis, B.; Demir, A.S. Direct enantioselectivealdol reactions catalyzed by a proline–thiourea host–guest complex. Chem. Commun. 2009, 9, 1088–1090.
- Poe, S.L.; Bogdan, A.R.; Mason, B.P.; Steinbacher, J.L.; Opalka, S.M.; McQuade, D.T. Use of Bifunctional Ureas to Increase the Rate of Proline-Catalyzed α-Aminoxylations. J. Org. Chem. 2009, 74, 1574–1580.
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480.
- Gómez Arrayás, R.; Carretero, J.C. Catalytic asymmetric direct Mannich reaction: A powerful tool for the synthesis of α,β-diamino acids. Chem. Soc. Rev. 2009, 38, 1940–1948.
- Verkade, J.M.M.; van Hemert, L.J.C.; Quaedfliegb, P.J.L.M.; Rutjes, F.P.J.T. Organocatalysed asymmetric Mannich reactions. Chem. Soc. Rev. 2008, 37, 29–41.
- Cai, X.-H.; Guo, H.; Bing, X. Recent progress in the asymmetric Mannich reaction. Eur. J. Chem. 2012, 3, 258–266.
- Duthaler, R.O. Proline-Catalyzed Asymmetric α-Amination of Aldehydes and Ketones—An Astonishingly Simple Access to Optically Active α-Hydrazino Carbonyl Compounds. Angew. Chem. Int. Ed. 2003, 42, 975–978.
- Merino, P.; Tejero, T. Organocatalyzed Asymmetric α-Aminoxylation of Aldehydes and Ketones—An Efficient Access to Enantiomerically Pure α-Hydroxycarbonyl Compounds, Diols, and Even Amino Alcohols. Angew. Chem. Int. Ed. 2004, 43, 2995–2997.
- Albrecht, Ł.; Jiang, H.; Jørgensen, K.A. Hydrogen-Bonding in Aminocatalysis: From Proline and Beyond. Chem. Eur. J. 2014, 20, 358–368.
- Liu, X.; Lin, L.; Feng, X. Amide-based bifunctional organocatalysts in asymmetric reactions. Chem. Commun. 2009, 41, 6145–6158.
- Tang, Z.; Jiang, F.; Yu, L.T.; Cui, X.; Gong, L.Z.; Mi, A.Q.; Jiang, Y.Z. Novel Small Organic Molecules for a Highly Enantioselective Direct Aldol Reaction. J. Am. Chem. Soc. 2003, 125, 5262–5263.
- Tang, Z.; Jiang, F.; Cui, X.; Gong, L.-Z.; Mi, A.-Q.; Jiang, Y.-Z.; Wu, Y.-D. Enantioselective direct aldol reactions catalyzed by l-prolinamide derivatives. Proc. Natl. Acad. Sci. USA 2004, 101, 5755–5760.
- Tang, Z.; Yang, Z.H.; Chen, X.H.; Cun, L.F.; Mi, A.Q.; Jiang, Y.Z.; Gong, L.Z. A Highly Efficient Organocatalyst for Direct Aldol Reactions of Ketones with Aldedydes. J. Am. Chem. Soc. 2005, 127, 9285–9289.
- Yang, H.; Carter, R.G. Proline Sulfonamide Based Organocatalysis: Better Late than Never. Synlett 2010, 19, 2827–2838.
- Berkessel, A.; Koch, B.; Lex, J. Proline-Derived N-Sulfonylcarboxamides: Readily Available, Highly Enantioselective and Versatile Catalysts for Direct Aldol Reactions. Adv. Synth. Catal. 2004, 346, 1141–1146.
- Cobb, A.J.A.; Shaw, D.M.; Longbottom, D.A.; Gold, J.B.; Ley, S.V. Organocatalysis with proline derivatives: Improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org. Biomol. Chem. 2005, 3, 84–96.
- Bellis, E.; Kokotos, G. 4-Substituted prolines as organocatalysts for aldol reactions. Tetrahedron 2005, 61, 8669–8676.
- Silva, F.; Sawicki, M.; Gouverneur, V. Enantioselective Organocatalytic Aldol Reaction of Ynones and Its Synthetic Applications. Org. Lett. 2006, 8, 5417–5419.
- Sundén, H.; Dahlin, N.; Ibrahem, I.; Adolfsson, H.; Cordova, A. Novel organic catalysts for the direct enantioselective α-oxidation of carbonyl compounds. Tetrahedron Lett. 2005, 46, 3385–3389.
- Wang, W.; Wang, J.; Li, H. A Simple and Efficient l-Prolinamide-Catalyzed α-Selenenylation Reaction of Aldehydes. Org. Lett. 2004, 6, 2817–2820.
- Gryko, D.; Chromiński, M.; Pielacińska, D.J. Prolinethioamides versus Prolinamides in Organocatalyzed Aldol Reactions—A Comparative Study. Symmetry 2011, 3, 265–282.
- Gryko, D.; Lipinski, R. l-Prolinethioamides—Efficient Organocatalysts for the Direct Asymmetric Aldol Reaction. Adv. Synth. Catal. 2005, 347, 1948–1952.
- Almaşi, D.; Alonso, D.A.; Nájera, C. Prolinamides versus Prolinethioamides as Recyclable Catalysts in the Enantioselective Solvent-Free Inter- and Intramolecular Aldol Reactions. Adv. Synth. Catal. 2008, 350, 2467–2472.
- Wang, B.; Chen, G.; Liu, L.; Chang, W.; Li, J. A Novel Proline-Valinol Thioamide Small Organic Molecule for a Highly Enantioselective Direct Aldol Reaction. Adv. Synth. Catal. 2009, 351, 2441–2448.
- Torii, H.; Nakadai, M.; Ishihara, K.; Saito, S.; Yamamoto, H. Asymmetric direct aldol reaction assisted by water and a proline-derived tetrazole catalyst. Angew. Chem. Int. Ed. 2004, 43, 1983–1986.
- Momiyama, N.; Torii, H.; Saito, S.; Yamamoto, H. O-nitroso aldol synthesis: Catalytic enantioselective route to α-aminooxy carbonyl compounds via enamine intermediate. Proc. Natl. Acad. Sci. USA 2004, 101, 5374–5378.
- Cobb, A.J.A.; Shaw, D.M.; Ley, S.V. 5-Pyrrolidin-2-yltetrazole: A New, Catalytic, More Soluble Alternative to Proline in an Organocatalytic Asymmetric Mannich-type Reaction. Synlett 2004, 3, 558–560.
- Hartikka, A.; Arvidsson, P.I. 5-(Pyrrolidine-2-yl)tetrazole: Rationale for the Increased Reactivity of the Tetrazole Analogue of Proline in Organocatalyzed Aldol Reactions. Eur. J. Org. Chem. 2005, 2005, 4287–4295.
- Maji, B.; Yamamoto, H. Proline-Tetrazole-Catalyzed Enantioselective N-Nitroso Aldol Reaction of Aldehydes with In Situ Generated Nitrosocarbonyl Compounds. Angew. Chem. Int. Ed. 2014, 53, 8714.
- Hayashi, Y.; Aratake, S.; Okano, T.; Takahashi, J.; Sumiya, T.; Shoji, M. Combined Proline–Surfactant Organocatalyst for the Highly Diastereo- and Enantioselective Aqueous Direct Cross-Aldol Reaction of Aldehydes. Angew. Chem. Int. Ed. 2006, 45, 5527–5529.
- Zlotin, S.G. Hydroxyproline Derivatives as Asymmetric Organocatalysts. In Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 1; Chapter 10; RSC Publishing: Cambridge, UK, 2015; p. 236.
- Martin, H.J.; List, B. Mining Sequence Space for Asymmetric Aminocatalysis: N-Terminal Prolyl-Peptides Efficiently Catalyze Enantioselective Aldol and Michael Reactions. Synlett 2003, 12, 1901–1902.
- Hernandez, J.G.; Juaristi, E. Asymmetric Aldol Reaction Organocatalyzed by (S)-Proline-Containing Dipeptides: Improved Stereoinduction under Solvent-Free Conditions. J. Org. Chem. 2011, 76, 1464–1467.
- Bisticha, A.; Triandafillidi, I.; Kokotos, C.G. tert-Butyl esters of peptides as organocatalysts for the asymmetric aldol reaction. Tetrahedron Asymmetry 2015, 26, 102–108.
- Lei, M.; Shi, L.; Li, G.; Chen, S.; Fang, W.; Ge, Z.; Cheng, T.; Li, R. Dipeptide-catalyzed direct asymmetric aldol reactions in the presence of water. Tetrahedron 2007, 63, 7892–7898.
- Agarwal, J. Progress in aminosugar derived asymmetric organocatalysis. Org. Biomol. Chem. 2016, 14, 10747–10762.
- Yeboah, E.M.O.; Yeboah, S.O.; Singh, G.S. Recent applications of Cinchona alkaloids and their derivatives as catalysts in metal-free asymmetric synthesis. Tetrahedron 2011, 67, 1725–1762.
- Chen, J.-R.; An, X.-L.; Zhu, X.-Y.; Wang, X.-F.; Xiao, W.-J. Rational Combination of Two Privileged Chiral Backbones: Highly Efficient Organocatalysts for Asymmetric Direct Aldol Reactions between Aromatic Aldehydes and Acylic Ketones. J. Org. Chem. 2008, 73, 6006–6009.
- Barrulas, P.; Benaglia, M.; Burke, A.J. Synthesis of novel cinchona-amino acid hybrid organocatalysts for asymmetric catalysis. Tetrahedron Asymmetry 2014, 25, 923–935.
- Guo, H.-M.; Cheng, L.; Cun, L.-F.; Gong, L.-Z.; Mi, A.-Q.; Jiang, Y.-Z. l-Prolinamide-catalyzed direct nitroso aldol reactions of α-branched aldehydes: A distinct regioselectivity from that with l-proline. Chem. Commun. 2006, 429–431.
- Saito, S.; Yamamoto, H. Design of Acid−Base Catalysis for the Asymmetric Direct Aldol Reaction. Acc. Chem. Res. 2004, 37, 570–579.
- Chen, J.R.; Lu, H.H.; Li, X.Y.; Cheng, L.; Wan, J.; Xiao, W.J. Readily Tunable and Bifunctional l-Prolinamide Derivatives: Design and Application in the Direct Enantioselective Aldol Reactions. Org. Lett. 2005, 7, 4543–4545.
- Gandhi, S.; Singh, V.K. Synthesis of Chiral Organocatalysts derived from Aziridines: Application in Asymmetric Aldol Reaction. J. Org. Chem. 2008, 73, 9411–9416.
- Banon-Caballero, A.; Guillena, G.; Nájera, C. Solvent-free direct enantioselective aldol reaction using polystyrene-supported N-sulfonyl-(Ra)-binam-d-prolinamide as a catalyst. Green Chem. 2010, 12, 1599–1606.
- Bradshaw, B.; Etxebarria-Jardi, G.; Bonjoch, J.; Viozquez, S.F.; Guillena, G.; Nájera, C. Efficient Solvent-Free Robinson Annulation Protocols for the Highly Enantioselective Synthesis of the Wieland–Miescher Ketone and Analogues. Adv. Synth. Catal. 2009, 351, 2482–2490.
- Viozquez, S.F.; Guillena, G.; Nájera, C.; Bradshaw, B.; Etxebarria-Jardi, G.; Bonjoch, J. (Sa,S)-N-pyrrolidine-2-Carboxamide: An Organocatalyst for the Direct Aldol Reaction. Org. Synth. 2011, 88, 317–329.
- Connon, S.J. Organocatalysis Mediated by (Thio)urea Derivatives. Chem. Eur. J. 2006, 12, 5418–5427.
- Tsakos, M.; Kokotos, C.G. Primary and secondary amine-(thio)ureas and squaramides and their applications in asymmetric organocatalysis. Tetrahedron 2013, 69, 10199–10222.
- Fotaras, S.; Kokotos, C.G.; Tsandi, E.; Kokotos, G. Prolinamides Bearing Thiourea Groups as Catalysts for Asymmetric Aldol Reactions. Eur. J. Org. Chem. 2011, 2011, 1310–1317.
- Kokotos, C.G. Construction of Tertiary Alcohols Bearing Perfluoroalkyl Chains Catalyzed by Prolinamide-Thioureas. J. Org. Chem. 2012, 77, 1131–1135.
- Narayaperumal, S.; Rivera, D.G.; Silva, R.C.; Paixao, M.W. Terpene-Derived Bifunctional Thioureas in Asymmetric Organocatalysis. ChemCatChem. 2013, 5, 2756–2773.
- Metrano, A.J.; Chinn, A.J.; Shugrue, C.R.; Stone, A.; Kim, B.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev. 2020, 120, 11479–11615.
- Wennemers, H. Asymmetric catalysis with peptides. Chem. Commun. 2011, 47, 12036–12041.
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev. 2007, 107, 5759–5812.
- Rodriguez-Llansola, F.; Miravet, J.F.; Escuder, B. A supramolecular hydrogel as a reusable heterogeneous catalyst for the direct aldol reaction. Chem. Commun. 2009, 47, 7303–7305.
- Hu, X.-M.; Zhang, D.-X.; Zhang, S.-Y.; Wang, P.-A. Highly modular dipeptide-like organocatalysts for direct asymmetric aldol reactions in brine. RSC Adv. 2015, 5, 39557–39564.
- Krattiger, P.; Kovasy, R.; Revell, J.D.; Ivan, S.; Wennemers, H. Increased Structural Complexity Leads to Higher Activity: Peptides as Efficient and Versatile Catalysts for Asymmetric Aldol Reactions. Org. Lett. 2005, 7, 1101–1103.
- D’Elia, V.; Zwicknagl, H.; Reiser, O. Short α/β-Peptides as Catalysts for Intra- and Intermolecular Aldol Reactions. J. Org. Chem. 2008, 73, 3262–3265.
- Agirre, M.; Arrieta, A.; Arrastia, I.; Cossio, F.P. Organocatalysts Derived from Unnatural α-Amino Acids: Scope and Applications. Chem. Asian J. 2019, 14, 44–66.
- List, B.; Coric, I.; Grygornko, O.O.; Kaib, P.S.J.; Komarov, I.; Lee, A.; Leutzsch, M.; Pan, S.C.; Tymtsunik, A.V.; van Gemmeren, M. The Catalytic Asymmetric α-Benzylation of Aldehydes. Angew. Chem. Int. Ed. 2014, 53, 282–285.
- Zhang, H.; Mitsumori, S.; Utsumi, N.; Imai, M.; Garcia-Delgado, N.; Mifsud, M.; Albertshofer, K.; Cheong, P.H.-Y.; Houk, K.N.F.; Tanaka, F.; et al. Catalysis of 3-Pyrrolidinecarboxylic Acid and Related Pyrrolidine Derivatives in Enantioselective anti-Mannich-Type Reactions: Importance of the 3-Acid Group on Pyrrolidine for Stereocontrol. J. Am. Chem. Soc. 2008, 130, 875–886.
- Gómez-Bengoa, E.; Maestro, M.; Mielgo, A.; Otazo, I.; Palomo, C.; Velilla, I. A 4-Hydroxypyrrolidine-Catalyzed Mannich Reaction of Aldehydes: Control of anti-Selectivity by Hydrogen Bonding Assisted by Brønsted Acids. Chem. Eur. J. 2010, 16, 5333–5342.
- Martín-Rapún, R.; Fan, X.; Sayalero, S.; Bahramnejad, M.; Cuevas, F.; Pericàs, M.A. Highly Active Organocatalysts for Asymmetric anti-Mannich Reactions. Chem. Eur. J. 2011, 17, 8780–8783.
- Chuan, Y.-M.; Chen, G.-H.; Gao, J.-Z.; Zhang, H.; Peng, Y.-G. A facile direct anti-selective catalytic asymmetric Mannich reaction of aldehydes with preformed N-Boc and N-Cbz imines. Chem. Commun. 2011, 47, 3260–3262.
- Gao, J.; Chuan, Y.; Li, J.; Xie, F.; Peng, Y. A convenient and mild chromatography-free method for the purification of the products of Wittig and Appel reactions. Org. Biomol. Chem. 2012, 10, 3730–3737.
- Kano, T.; Maruoka, K. Unique properties of chiral biaryl-based secondary aminecatalysts for asymmetric enamine catalysis. Chem. Sci. 2013, 4, 907–915.
- Kano, T.; Yamaguchi, Y.; Tokuda, O.; Maruoka, K. anti-Selective Direct Asymmetric Mannich Reactions Catalyzed by Axially Chiral Amino Sulfonamide as an Organocatalyst. J. Am. Chem. Soc. 2005, 127, 14609–16408.
- Kano, T.; Yamaguchi, Y.; Maruoka, K. A Designer Axially Chiral Amino Sulfonamide as an Efficient Organocatalyst for Direct Asymmetric Mannich Reactions of N-Boc-Protected Imines. Angew. Chem. Int. Ed. 2009, 48, 1838–1840.
- Kano, T.; Yamaguchi, Y.; Tanaka, Y.; Maruoka, K. syn-Selective and Enantioselective Direct Cross-Aldol Reactions between Aldehydes Catalyzed by an Axially Chiral Amino Sulfonamide. Angew. Chem. Int. Ed. 2007, 46, 1768–1770.
- Kano, T.; Takai, J.; Tokuda, O.; Maruoka, K. Design of an Axially Chiral Amino Acid with a Binaphthyl Backbone as an Organocatalyst for a Direct Asymmetric Aldol Reaction. Angew. Chem. Int. Ed. 2005, 44, 3055–3057.
- Kano, T.; Yamaguchi, Y.; Maruoka, K. A Designer Axially Chiral Amino Sulfonamide as an Efficient Organocatalyst for Direct Asymmetric anti-Selective Mannich Reactions and syn-Selective Cross-Aldol Reactions. Chem. Eur. J. 2009, 15, 6678–6687.
- Xu, L.-W.; Luo, J.; Lu, Y. Asymmetric catalysis with chiral primary amine-based organocatalysts. Chem. Commun. 2009, 14, 1807–1821.
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071.
- Cordova, A.; Zou, W.; Ibrahem, I.; Reyes, E.; Engqvist, M.; Liao, W.-W. Acyclic amino acid-catalyzed direct asymmetric aldol reactions: Alanine, the simplest stereoselective organocatalyst. Chem. Commun. 2005, 28, 3586–3588.
- Xu, L.-W.; Lu, Y. Primary amino acids: Privileged catalysts in enantioselective organocatalysis. Org. Biomol. Chem. 2008, 6, 2047–2053.
- Liu, J.; Yang, Z.; Wang, Z.; Wang, F.; Chen, X.; Liu, X.; Feng, X.; Su, Z.; Hu, C. Asymmetric Direct Aldol Reaction of Functionalized Ketones Catalyzed by Amine Organocatalysts Based on Bispidine. J. Am. Chem. Soc. 2008, 130, 5654–5655.
- Nakayama, K.; Maruoka, K. Complete Switch of Product Selectivity in Asymmetric Direct Aldol Reaction with Two Different Chiral Organocatalysts from a Common Chiral Source. J. Am. Chem. Soc. 2008, 130, 17666–17667.
- Deng, Y.H.; Chen, J.-Q.; He, L.; Kang, T.-R.; Liu, Q.-Z.; Luo, S.-W.; Yuan, W.-C. Highly Enantioselective Aldol Reactions between Acetaldehyde and Activated Acyclic Ketones Catalyzed by Chiral Primary Amines. Chem. Eur. J. 2013, 19, 7143–7150.
- Dziedzic, P.; Zou, W.; Hafren, J.; Cordova, A. The small peptide-catalyzed direct asymmetric aldol reaction in water. Org. Biomol. Chem. 2006, 4, 38–40.
- Wu, F.-C.; Da, C.-S.; Du, Z.-X.; Guo, Q.-P.; Li, W.-P.; Yi, L.; Jia, Y.-N.; Ma, X. N-Primary-Amine-Terminal β-Turn Tetrapeptides as Organocatalysts for Highly Enantioselective Aldol Reaction. J. Org. Chem. 2009, 74, 4812–4818.
- Du, Z.-H.; Tao, B.-X.; Yuan, M.; Qin, W.-J.; Xu, Y.-L.; Wang, P.; Da, C.-S. Peptide-Catalyzed Highly Asymmetric Cross-Aldol Reaction of Aldehydes to Biomimetically Synthesize 1,4-Dicarbonyls. Org. Lett. 2020, 22, 4444–4450.
- Liu, T.-Y.; Cui, H.-L.; Jiang, K.; Du, W.; He, Z.-Q.; Chen, Y.-C. Organocatalytic and Highly Enantioselective Direct α-Amination of Aromatic Ketones. Org. Lett. 2007, 9, 3671–3674.
- Reyes, E.; Uria, U.; Carrillo, L.; Vicario, J.L. The Catalytic Enantioselective Michael reaction. In Organic Reactions; Denmark, S.E., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 1–898.
- List, B.; Pojarliev, P.; Martin, H.J. Efficient Proline-Catalyzed Michael Additions of Unmodified Ketones to Nitro Olefins. Org. Lett. 2001, 3, 2423–2425.
- Cobb, A.J.A.; Longbottom, D.A.; Shaw, D.M.; Ley, S.V. 5-Pyrrolidin-2-yltetrazole as an asymmetric organocatalyst for the addition of ketones to nitro-olefins. Chem. Commun. 2004, 16, 1808–1809.
- Mitchell, C.E.T.; Cobb, A.J.A.; Ley, S.V. A Homo-Proline Tetrazole as an Improved Organocatalyst for the Asymmetric Michael Addition of Carbonyl Compounds to Nitro-Olefins. Synlett 2005, 4, 611–614.
- Cao, C.-L.; Ye, M.-C.; Sun, X.-L.; Tang, Y. Pyrrolidine−Thiourea as a Bifunctional Organocatalyst: Highly Enantioselective Michael Addition of Cyclohexanone to Nitroolefins. Org. Lett. 2006, 8, 2901–2904.
- Kokotos, C.G.; Limnios, D.; Triggidou, D.; Trifonidou, M.; Kokotos, G. Novel pyrrolidine-thiohydantoins/thioxotetrahydropyrimidinones as highly effective catalysts for the asymmetric Michael addition. Org. Biomol. Chem. 2011, 9, 3386–3395.
- Mahato, C.K.; Mukherjee, S.; Kundu, M.; Pramanik, A. Pyrrolidine-Oxadiazolone Conjugates as Organocatalysts in Asymmetric Michael Reaction. J. Org. Chem. 2019, 84, 1053–1063.
- Kucherenko, A.S.; Lisnyak, V.G.; Kostenko, A.A.; Kochetkov, S.V.; Zlotin, S.G. C 2-Symmetric pyrrolidine-derived squaramides as recyclable organocatalysts for asymmetric Michael reactions. Org. Biomol. Chem. 2016, 14, 9751–9759.
- Kaur, A.; Singh, K.N.; Sharma, E.; Rani, S.P.; Sharma, S.K. Pyrrolidine-carbamate based new and efficient chiral organocatalyst for asymmetric Michael addition of ketones to nitroolefins. Tetrahedron 2018, 74, 6137–6143.
- Reyes-Rangel, G.; Vargas-Caporali, J.; Juaristi, E. Asymmetric Michael addition reaction organocatalyzed by stereoisomeric pyrrolidine sulfinamides under neat conditions. A brief study of self-disproportionation of enantiomers. Tetrahedron 2017, 73, 4707–4718.
- Cruz-Hernández, C.; Martínez-Martínez, E.; Hernández-González, P.E.; Juaristi, E. Synthesis of a New N-Diaminophosphoryl-N′-thiourea as a Chiral Organocatalyst for the Stereoselective Michael Addition of Cyclohexanone to Nitrostyrenes and Chalcones—Application in Cascade Processes for the Synthesis of Polycyclic Systems. Eur. J. Org. Chem. 2018, 2018, 6890–6900.
- Palomo, C.; Vera, S.; Mielgo, A.; Gomez-Bengoa, E. Highly Efficient Asymmetric Michael Addition of Aldehydes to Nitroalkenes Catalyzed by a Simple trans-4-Hydroxyprolylamide. Angew. Chem. Int. Ed. 2006, 45, 5984–5987.
- Wiesner, M.; Upert, G.; Angelici, G.; Wennemers, H. Enamine Catalysis with Low Catalyst Loadings—High Efficiency via Kinetic Studies. J. Am. Chem. Soc. 2010, 132, 6–7.
- Wiesner, M.; Revell, J.D.; Tonazzi, S.; Wennemers, H. Peptide Catalyzed Asymmetric Conjugate Addition Reactions of Aldehydes to Nitroethylene—A Convenient Entry into γ2-Amino Acids. J. Am. Chem. Soc. 2008, 130, 5610–5611.
- Duschmale, J.; Wennemers, H. Adapting to Substrate Challenges: Peptides as Catalysts for Conjugate Addition Reactions of Aldehydes to α,β-Disubstituted Nitroolefins. Chem. Eur. J. 2012, 18, 1111–1120.
- Kastl, R.; Wennemers, H. Peptide-Catalyzed Stereoselective Conjugate Addition Reactions Generating All-Carbon Quaternary Stereogenic Centers. Angew. Chem. Int. Ed. 2013, 52, 7228–7232.
- Wiesner, M.; Revell, J.D.; Wennemers, H. Tripeptides as Efficient Asymmetric Catalysts for 1,4-Addition Reactions of Aldehydes to Nitroolefins–A Rational Approach. Angew. Chem. Int. Ed. 2008, 47, 1871–1874.
- Wiesner, M.; Neuburger, M.; Wennemers, H. Tripeptides of the Type H-D-Pro-Pro-Xaa-NH2 as Catalysts for Asymmetric 1,4-Addition Reactions: Structural Requirements for High Catalytic Efficiency. Chem. Eur. J. 2009, 15, 10103–10109.
- Bachle, F.; Duschmale, J.; Ebner, C.; Pfaltz, A.; Wennemers, H. Organocatalytic Asymmetric Conjugate Addition of Aldehydes to Nitroolefins: Identification of Catalytic Intermediates and the Stereoselectivity-Determining Step by ESI-MS. Angew. Chem. Int. Ed. 2013, 52, 12619–12623.
- Schnitzer, T.; Wennemers, H. Influence of the Trans/Cis Conformer Ratio on the Stereoselectivity of Peptidic Catalysts. J. Am. Chem. Soc. 2017, 139, 15356–15362.
- Schnitzer, T.; Wennemers, H. Effect of γ-Substituted Proline Derivatives on the Performance of the Peptidic Catalyst H-dPro-Pro-Glu-NH2. Synthesis 2018, 50, 4377–4382.
- Siebler, C.; Maryasin, B.; Kuemin, M.; Erdmann, R.S.; Rigling, C.; Grunenfelder, C.; Ochsenfeld, C.; Wennemers, H. Importance of dipole moments and ambient polarity for the conformation of Xaa–Pro moieties—A combined experimental and theoretical study. Chem. Sci. 2015, 6, 6725–6730.
- Rigling, C.; Kisunzu, J.K.; Duschmale, J.; Haussinger, D.; Wiesner, M.; Ebert, M.-O.; Wennemers, H. Conformational Properties of a Peptidic Catalyst: Insights from NMR Spectroscopic Studies. J. Am. Chem. Soc. 2018, 140, 10829–10838.
- Duschmale, J.; Wiest, J.; Wiesner, M.; Wennemers, H. Effects of internal and external carboxylic acids on the reaction pathway of organocatalytic 1,4-addition reactions between aldehydes and nitroolefins. Chem. Sci. 2013, 4, 1312–1318.
- Schnitzer, T.; Mohler, J.S.; Wennemers, H. Effect of the enamine pyramidalization direction on the reactivity of secondary amine organocatalysts. Chem. Sci. 2020, 11, 1943–1947.
- Bures, J.; Armstrong, A.; Blackmond, D.G. Explaining Anomalies in Enamine Catalysis: “Downstream Species” as a New Paradigm for Stereocontrol. Acc. Chem. Res. 2016, 49, 214–222.
- Znabet, A.; Ruijter, E.; de Kanter, F.J.J.; Köhler, V.; Helliwell, M.; Turner, N.J.; Orru, R.V.A. Highly Stereoselective Synthesis of Substituted Prolyl Peptides Using a Combination of Biocatalytic Desymmetrization and Multicomponent Reactions. Angew. Chem. Int. Ed. 2010, 49, 5289–5292.
- Cortes-Clerget, M.; Gager, O.; Monteil, M.; Pirat, J.-L.; Migianu-Griffoni, E.; Deschamp, J.; Lecouvey, M. Novel Easily Recyclable Bifunctional Phosphonic Acid Carrying Tripeptides for the Stereoselective Michael Addition of Aldehydes with Nitroalkenes. Adv. Synth. Catal. 2016, 358, 34–40.
- Durini, M.; Sahr, F.A.; Kuhn, M.; Civera, M.; Gennari, C.; Piarulli, U. Bifunctional 2,5-Diketopiperazines as Efficient Organocatalysts for the Enantioselective Conjugate Addition of Aldehydes to Nitroolefins. Eur. J. Org. Chem. 2011, 2011, 5599–5607.
- Tsogoeva, S.B.; Jagtap, S. Dual Catalyst Control in the Chiral Diamine-Dipeptide-Catalyzed Asymmetric Michael Addition. Synlett 2004, 14, 2624–2626.
- de la Torre, A.F.; Rivera, D.G.; Ferreira, M.A.B.; Correia, A.G.; Paixao, M.W. Multicomponent Combinatorial Development and Conformational Analysis of Prolyl Peptide–Peptoid Hybrid Catalysts: Application in the Direct Asymmetric Michael Addition. J. Org. Chem. 2013, 78, 10221–10232.
- Schnitzer, T.; Budinská, A.; Wennemers, H. Organocatalysed conjugate addition reactions of aldehydes to nitroolefins with anti selectivity. Nat. Catal. 2020, 3, 143–147.
- Tsogoeva, S.B.; Wei, S. Highly enantioselective addition of ketones to nitroolefins catalyzed by new thiourea–amine bifunctional organocatalysts. Chem. Commun. 2006, 13, 1451–1453.
- Yalalov, D.A.; Tsogoeva, S.B.; Schmatz, S. Chiral Thiourea-Based Bifunctional Organocatalysts in the Asymmetric Nitro-Michael Addition: A Joint Experimental-Theoretical Study. Adv. Synth. Catal. 2006, 348, 826–832.
- Desmarchelier, A.; Coeffard, V.; Moreau, X.; Greck, C. Asymmetric organocatalytic functionalization of α,α-disubstituted aldehydes through enamine activation. Tetrahedron 2014, 70, 2491–2513.
- Lalonde, M.P.; Chen, Y.; Jacobsen, E.N. A Chiral Primary Amine Thiourea Catalyst for the Highly Enantioselective Direct Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes. Angew. Chem. Int. Ed. 2006, 45, 6366–6370.
- Huang, H.; Jacobsen, E.N. Highly Enantioselective Direct Conjugate Addition of Ketones to Nitroalkenes Promoted by A Chiral Primary Amine−Thiourea Catalyst. J. Am. Chem. Soc. 2006, 128, 7170–7171.
More