Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alison Allan and Version 2 by Fanny Huang.
One of the common locations for breast cancer metastasis is the lung, which is associated with significant morbidity and mortality. Several studies have demonstrated that breast cancer-derived extracellular vesicles secreted from the primary breast tumor play a key role in establishing the lung pre-metastatic niche to support colonization of metastatic tumor cells.
- breast cancer
- lung metastasis
- extracellular vesicles
1. Extracellular Vesicles
Extracellular vesicles are lipid-bound vesicles secreted by all cell types. Originally believed to be cellular debris, further analysis has revealed that EVs function as packaging entities containing molecular cargo such as proteins, nucleic acids, lipids, and metabolites, which can be transported through the body and transferred between cells as a mode of communication [1][27]. Extracellular vesicles are classified based on size, function, and biogenesis [2][28]. Exosomes (30–150 nm) are intraluminal vesicles that form from the plasma membrane via the endosomal pathway and have a regulated secretion [3][4][29,30]. Endosomes (100–500 nm) are degradative, pre-lysosomal vesicles that primarily function in recycling or degradative systems of ligands and macromolecules [5][6][31,32]. Early sorting endosomes (ESE) are initially formed by inward invagination of the plasma membrane, followed by maturation into late sorting endosomes (LSE), and a second invagination of the LSE to generate multivesicular bodies (MVB) [7][33]. Multivesicular bodies are a subset of endosomal vesicles that can be transported to the plasma membrane via cytoskeletal and microtubule networks. Multivesicular bodies are generated by inward invagination of the endosomal membrane with the release of discrete intraluminal vesicles (ILVs) into the MVB lumen. The MVBs can then either fuse with lysosomes and be degraded, or fuse with the plasma membrane and release the contained ILVs in the extracellular space as exosomes. Deriving from ILVs, exosomes have size constraints and cannot be larger than 150–200 nm, depending also on the method used to detect the size [2][7][28,33]. Microvesicles (30–1000 nm) are produced and released directly into the extracellular space via outward blebbing of the plasma membrane instigated by increased cytosolic calcium levels. Activation of calcium-dependent proteases such as calpain results in disruption of the cytoskeleton and subsequent vesicle release from domains called lipid rafts [3][8][29,34]. Microvesicles have selectively enriched contents and (similar to exosomes) have a regulated release, making them important signaling components in response to physiological changes [9][10][35,36]. Apoptotic bodies (50–5000 nm) are formed from dying cells as they degrade their cellular content [3][11][29,37]. They are formed via apoptotic cell disassembly, a general three-step process requiring plasma membrane blebbing, outward membrane protrusions, and fragmentation into EVs [12][38]. The mechanism of vesicle formation varies depending on regulators involved [13][14][39,40]. The release of apoptotic bodies serves as a mode of clearing dead cell debris via phagocytes and communicating with surrounding cells via the uptake of encapsulated proteins and nucleic acids. Finally, large oncosomes (1–10 μm) are exclusively cancer-related EVs. They are generated by the blebbing of amoeboid cancers and have been identified in luminal A and triple-negative breast cancer as well as in prostate cancer [15][16][41,42]. Unlike microvesicles and exosomes which have been the focus of more research, the full extent of apoptotic body and large oncosome signaling and functionality has not yet been elucidated [12][17][38,43].
A growing body of research highlights the importance of EV signaling in breast cancer progression, particularly in the context of the PMN and the metastatic microenvironment. This signaling is mediated in large part by the cargo contained within the EVs. As an example, in addition to proteins, EVs contain a variety of non-coding RNAs including circular RNAs (circRNAs), microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). Along with other cargo components, non-coding RNAs contained within EVs contribute to many important aspects of the metastatic process, such as angiogenesis, cancer–stromal cell communication, and epithelial-to-mesenchymal transition (EMT) [18][19][44,45]. Although lipids are important in regulating EV contents and EV-derived lipids can modify recipient cell activity [20][46], to date there is currently limited data in the literature focused on the role of lipid-based EV cargo in influencing breast cancer features. While work by Nishida-Aoki and colleagues has demonstrated that highly metastatic breast cancer EV lipid contents can activate angiogenesis upon uptake by endothelial cells [21][47], further research in this important area is needed.
Relative to the normal physiological state, the process of tumorigenesis has an impact on EV release, both in terms of the number of EVs released as well as their cargo/composition. In particular, the production and packaging of EVs is influenced by the conditions of the individual and various environmental stimuli. Cargo within EVs can be influenced by conditions such as obesity, cardiovascular disease, and autoimmune disorders to further support tumorigenesis. For example, EVs derived from obese adipose tissue promoted the proliferation of MDA-MB-231 and MCF7 breast cancer cells along with increasing invasion and migration of MDA-MB-231 cells. This was accomplished through activation of extracellular signal-regulated kinase (ERK) and phosphatidylinositol-3-kinase (PI3K) pathways [22][48]. There have been associations discovered between cardiovascular disease and high levels of microvesicles that have pro-angiogenic effects and influence inflammation [23][49]. Furthermore, autoimmune organs can produce EVs that carry autoantigens which trigger unwanted immune responses to assist tumor cells in successful immune evasion [24][50].
Extracellular vesicles are released at higher rates in environments of high temperature, acidity, and hypoxia, a feature that is particularly relevant in cancer since tumors are inherently hypoxic [25][26][27][51,52,53]. Once a tumor grows beyond a 70–200 μm proximity from nearby blood vessels, cancer cells become oxygen-starved [28][54]. Furthermore, research into the disparity between hypoxic versus normoxic cancer EVs has revealed that hypoxia affects EV size, production, and contents [29][55]. The altered signaling that is observed during hypoxia is largely driven by hypoxia-inducible factors (HIFs), a protein family of oxygen homeostasis regulators. These transcription factors exist in three variants (HIF1, HIF2 and HIF3) with an α or β subunit. The β subunit is constitutively expressed and minimally reactive to oxygen saturation, while the α subunit is increasingly expressed under hypoxic conditions. Oxygen deprivation promotes the heterodimerization of HIFα and HIFβ, resulting in transcription factor functionality and increased EV release [29][30][55,56]. With respect to breast cancer, HIF-1α has been associated with EV release. Pachane et al. [31][57] compared the proteomes of EVs derived from MDA-MB-231 breast cancer cells grown in hypoxic versus normoxic conditions and observed that hypoxic EVs were enriched with proteins associated with the mTOR, TGF-β and pro-angiogenic VEGFA/VEGFR2 pathways. In breast cancer, mTOR activation contributes to increased proliferation, migration, and invasion; TGF-β signaling is involved in the differentiation of resident fibroblasts into cancer-associated fibroblasts, and endothelial uptake of VEGF promotes angiogenesis [32][33][34][35][58,59,60,61]. Overall, these studies support the concept that hypoxia is an important physiological regulator of breast cancer EVs, resulting in EV-mediated cancer–stromal interactions and cancer-cancer signaling that support tumor growth and metastasis.
2. Influence of Extracellular Vesicles on Lung Stromal Components
2.1. Endothelial Cells
One of the crucial stromal components in the lung is endothelial cells, which form a single layer lining along all blood vessels [36][93]. They are necessary for the process of angiogenesis—the formation of new blood vessels that supply nutrients and oxygen for tumor growth and also facilitate tumor cell dissemination [37][94]. Endothelial cells respond to pro- and anti-angiogenic factors released by the microenvironment that can either promote or inhibit vessel formation, respectively [36][93]. They also perform a barrier function, since substances in the blood are required to cross the endothelial layer in order to enter another site [36][93]. In the tumor microenvironment, the balance between these factors can be disrupted with an increase in pro-angiogenic factors to promote the formation of new vasculature [37][94]. Moreover, cancer-associated endothelial cells take on a different phenotype than that seen in normal tissues. In the normal physiological state, endothelial cells typically form organized and efficient vasculature with high integrity, allowing tightly regulated passage of nutrients to tissues and control of blood flow. In contrast, cancer-associated endothelial cells often form disorganized vasculature that is morphologically abnormal [38][95]. This vasculature tends to be unstable and leaky, influencing blood flow throughout the tumor [38][95] and supporting the metastatic process by easing the extravasation step in which tumor cells need to cross the endothelial monolayer to enter the secondary site [39][10].
There is growing evidence that tumor-derived EVs can promote angiogenesis by regulating the activity of endothelial cells at distant secondary sites to facilitate metastasis [28][40][41][42][43][54,96,97,98,99]. Zhou et al. demonstrated that EVs purified from triple-negative MDA-MB-231 breast cancer cells promoted metastasis-supporting behavior of primary human microvascular endothelial cells (HMVECs) [44][75]. They observed an increase in migration and permeability of the endothelial layer in vitro which was attributed to the presence of miR-105 in EVs derived from MDA-MB-231 cells [44][75]. The primary target of miR-105 is ZO-1, which was shown to be downregulated in HMVECs treated with EVs [44][75]. These results were replicated in vivo, where mice injected with MDA-MB-231 EVs showed similar pre-metastatic changes in lung endothelial cells, resulting in increased metastasis to the lung [44][75]. Another factor that regulates vessel permeability and contacts between endothelial cells are adherens junctions, specifically vascular endothelial cadherin (VE-cadherin) [45][74]. A study by Di Modica et al. showed that EVs from MDA-MB-231 cells were able to reduce the expression of VE-cadherin in endothelial cells, resulting in impaired endothelial function and enhanced permeability of the endothelial layer [45][74]. Endothelial cells treated with MDA-MB-231 EVs were found to display increased passage of breast cancer cells across the endothelium and a loss of cell-to-cell contacts in vitro [45][74]. This effect was attributed to the presence of miR-939 in these breast-cancer-derived EVs which targeted and disrupted expression of VE-cadherin [45][74].
In addition to miRNAs, growing evidence demonstrates that proteins contained within EVs can modify the behaviour of endothelial cells to promote angiogenic processes [46][72]. Specifically, breast-cancer-derived EVs have been shown to harbor annexin II (Anx-II), a protein associated with various cancer processes such as migration, proliferation, angiogenesis, and extracellular matrix degradation [46][72]. Maji and colleagues demonstrated that EVs from MCF10CA1a breast cancer cells contained Anx-II that resulted in increased endothelial tube formation in vitro and in vivo [46][72]. Moreover, when mice were injected with EVs from lung metastatic MDA-MB-4175 cells, the EVs localized primarily to the lung and resulted in higher numbers of lung metastases [46][72]. Another study comparing EVs from non-tumorigenic HME-1 cells to EVs from MDA-MB-231 cells demonstrated that MDA-MB-231 EVs were enriched in nucleoside diphosphate kinase (NDPK-B) expression and phosphotransferase activity [47][65]. Although NDPK-B is involved in several functions, its primary role is to transfer phosphate from nucleoside triphosphates to nucleoside disphosphates in nucleotide metabolism [48][100]. When endothelial cells were treated with NDPK-B+ EVs, enhanced migration and increased permeability of the endothelial monolayer was observed [47][65]. These changes were suggested to increase angiogenesis and extravasation in the lung to promote metastasis [47][65]. In mouse models, EV treatments showed an increase in pulmonary vascular leakage and greater lung metastasis that were caused by disruptions to the purinergic signaling pathway through changes in NDPK-B [47][65]. Together, these studies demonstrate the ability of breast-cancer-derived EVs to transport cargo to endothelial cells in the lung. By inducing pro-tumorigenic changes including increased permeability of the endothelial layer and enhanced angiogenesis, metastatic breast tumor cells are more likely to extravasate into the lung and colonize, leading to the successful formation of macrometastases.
2.2. Fibroblasts
Another important stromal component is fibroblasts, the primary source of ECM components in lung tissue. These cells generate optimal tissue conditions for lung function by synthesizing what is termed the “matrisome”. The matrisome is composed of all structural and adhesive proteins and ground substance components such as proteoglycans, glycoproteins, fibrillar proteins, and ECM-modifying proteins [49][101]. In addition, fibroblasts generate the basement membrane which separates the epithelium and surrounding stroma. This membrane serves as a barrier composed of type IV collagen and laminins with varying permeability, promoting the adhesion and migration of attaching cells via integrins and initiating cell signaling by releasing growth factors and other ECM-remodeling enzymes [50][51][102,103]. The ability of fibroblasts to create and remodel the lung ECM dictates interactions between all stromal cell components including endothelial cells, adipocytes, immune cells, neuronal cells, and others [52][104]. Additionally, the remodeling abilities of fibroblasts are essential for tissue repair during wound healing [53][105].
Thus far, the literature has emphasized the importance of fibroblast function in promoting cancer metastasis. The generation of heterogenous groups of cancer-associated fibroblasts (CAFs) has proven to be an essential step for remodeling the lung ECM into a cancer-promoting environment [54][106]. Research suggests that activation of the IL-6/STAT3, FGF2/FGR1, TGF-β/SMAD, and NF-κB signaling cascades may contribute to the activation and function of CAFs from normal fibroblasts [55][107]. Several studies have shown that during lung metastasis, there is significant crosstalk between primary breast tumor cells and lung fibroblasts that is mediated through EVs [56][108]. In breast cancer, specific RNAs and proteins contained within tumor-derived EVs such as miR-9, TGF-β, and Survivin activate CAFs [57][58][59][60][66,79,109,110]. Communication via EV release and uptake by fibroblasts supports the formation of a lung PMN and the establishment/growth of colonizing metastases.
Lung fibroblasts also support PMN formation by modulating the inflammasome and deposition of cancer-supportive ECM components. Hoshino and colleagues found that lung-targeted MDA-MB-231 EVs activated fibroblast S100 genes responsible for proliferation and migration [61][64]. In vivo studies have also shown that activation of S100A4 in mouse lung fibroblasts attracts T-lymphocytes which release cytokines that promote breast to lung metastasis. Deletion of S100A4+ fibroblasts have been shown to hinder metastasis, emphasizing the role of fibroblasts in inflammation and inflammatory cell recruitment [62][111]. Additional work by Gong et al. found that cyclooxygenase 2-expressing (COX-2+) resident lung fibroblasts promoted reprogramming of lung myeloid cells to reform the immune microenvironment. Deletion of the PTGS2 gene encoding COX2 prevented fibroblast-mediated immune remodeling and subsequently impaired breast cancer metastasis to the lungs [63][112]. To further understand how breast cancers generate inflammatory, cancer-associated fibroblasts (iCAFs), Gonzalez-Callejo and colleagues treated lung fibroblasts with MDA-MB-231 EVs, then re-treated MDA-MB-231 cells with conditioned media acquired from EV-treated fibroblasts. They reported that EV treatment induced increased IL-6, IL-8, and CXCL1 secretion in fibroblasts, which corresponded with the acquisition of chemoresistance to paclitaxel and the expression of Nanog and ALDH1A1 stem cell markers in MDA-MB-231 cells [64][113]. These findings support the generation of a metastasis-promoting lung inflammasome organized by fibroblast recruitment of EVs.
A hallmark of PMN formation is remodeling of lung ECM components. Medeiros et al. [65][114] observed that mice bearing triple-negative SUM159 breast tumors demonstrated enhanced levels of periostin, fibronectin, tenascin-c, matrix metalloproteinase 9, collagen A1, C-C chemokine ligand 2 (CCL2), and lysyl oxidase in the lung when compared to the lungs of tumor-naïve or MCF7 tumor-bearing mice. Additional in vitro work has confirmed that lung fibroblasts only demonstrate elevated periostin and fibronectin levels when treated with triple-negative breast cancer EVs, and not luminal A EVs [65][114]. Earlier work by Libring et al. found that breast cancer EVs greatly improved the deposition of aberrant fibronectin clusters by lung fibroblasts consistent with fibronectin patterns at the primary tumor [66][115]. Increased ECM deposition at the primary tumor and PMN stiffens tissues to promote breast to lung metastasis [67][68][69][116,117,118]. Studies by Hoshino et al. suggest that lung fibroblast EV uptake is dependent on the presence of integrin β4 (ITGβ4), which is predominantly expressed in lung-targeting, triple-negative cancers [61][64]. Together, these findings suggest that lung ECM remodeling is linked to organotropic breast cancer EVs that prime the lung microenvironment for metastasis.
Aside from establishing the PMN, CAFs are important in the tumor microenvironment for cancer progression and can release their own EVs that contribute to this. CAF-derived EVs promote breast cancer growth and tumorigenicity [56][108]. Cancer-associated fibroblast EVs containing miR-500a-5p were transferred to MDA-MB-231 and MCF7 breast cancer cells. This resulted in the downregulation of ubiquitin-specific peptidase 28 (USP28) and induced breast cancer cell proliferation, migration, invasion, and EMT [70][119]. Patient-derived CAF EVs have been shown to contain miR-21, miR-143, and miR-378e and to promote EMT and stemness in T47D breast cancer cells [71][120]. Additionally, CAFs can reprogram breast cancer metabolism to promote an aggressive phenotype. CAF EVs containing lncRNA SNHG3 attenuated miR-330-5p activity to promote pyruvate kinase M1/M2 (PKM) function in support of enhanced glycolysis and proliferation of cancer in vitro and in vivo [72][121]. Interestingly, MDA-MB-231 EVs enriched with ITGβ4 activated mitophagy and lactate generation in CAFs, and CAF-conditioned media used to re-treat MDA-MB-231 cells enhanced breast cancer invasiveness, proliferation, and EMT [73][122]. These findings highlight the importance of breast cancer and CAF-derived EVs in the lung microenvironment to support critical aspects of successful metastasis such as proliferation, invasion, and EMT.
2.3. Immune Cells
At each step of the metastatic cascade, it is vital that tumor cells evade immune destruction to successfully survive and reach a distant organ for colonization [74][11]. Even at the new site, immune components are altered to form an immunosuppressive environment that facilitates the creation of the PMN. In the lung, two specific populations of macrophages exist including alveolar macrophages found in the airway and alveolar lumen, and interstitial macrophages found in the parenchyma [75][123]. These macrophages are part of the innate immune system and take on different phenotypes depending on their environment [76][124]. Typically, M1 macrophages are pro-inflammatory, have enhanced antigen presentation and exert anti-tumorigenic effects, while M2 macrophages are anti-inflammatory and promote tissue remodeling and tumor progression [76][124]. It has been observed that EVs from MDA-MB-231 cells carry miR-138-5p, which is transferred to macrophages to decrease the expression of lysine demethylase 6B (KDM6B). This ultimately leads to M2 polarization and transcriptional inhibition of pro-inflammatory factors involved in M1 polarization [77][78]. When mice bearing breast tumors were engrafted with macrophages treated with miR-138-5p breast-cancer-derived EVs, significantly higher incidences of lung metastasis were observed, suggesting that miR-138-5p may signal lung macrophages to induce pre-metastatic changes to the tumor immune microenvironment [77][78]. Another study revealed that MDA-MB-231 cells that exhibit high signal-induced proliferation-associated 1 (SIPA1) protein expression can release EVs with upregulated expression of myosin-9 to promote the recruitment of macrophages and metastasis of tumor cells to the lung [78][68]. Neutrophils are another component of the innate immune system that can undergo changes to promote tumorigenesis [79][125]. It has been demonstrated that Lin28B, an RNA binding protein, increased breast cancer stem cell populations which are a main source of EVs with low let-7 miRNAs [80][90]. These EVs were able to recruit neutrophils and encourage M2 conversion to build an immunosuppressive PMN in the lung mediated by programmed death-ligand 2 (PD-L2) upregulation and cytokine imbalance [80][90].
With regard to adaptive immunity, T cells play a crucial role in anti-tumor immunity; however, their activity can be stunted depending on the microenvironment [81][126]. EVs derived from human MDA-MB-231 and mouse 4T1 breast cancer cells have been shown to express programmed death-ligand 1 (PD-L1) on their surface, which inhibits T cell activation and cytotoxic functions to assist tumor immune evasion [82][127]. It has also been reported that MDA-MB-231 cells can release and transfer the TGF-β type II receptor (TβRII) through EVs to recipient cells to trigger the TGF-β signaling pathway [34][60]. The EVs carrying TβRII promote CD8+ T cell exhaustion by stimulating activation of SMAD3 (mothers against decapentaplegic homolog 3) to associate with T cell factor 1 (TCF1) transcription factor while also promoting EMT [34][60]. Nude mice injected with 4T07 breast cancer cells experienced significantly higher lung metastasis and lower metastasis-free survival when treated with TβRII+ EVs. This demonstrates the ability of EVs to establish an immunosuppressive PMN in the lung [34][60].
Cytokines are secreted or membrane-bound proteins that allow for intercellular communication [83][128]. The set of cytokines present in the tumor microenvironment have been shown to influence the behaviour of immune cells while promoting or inhibiting cancer pathogenesis [83][128]. Murine 4T1 breast cancer cells release LC3+ EVs which stimulate lung fibroblasts to produce CCL2 through the TLR2/MyD88/NF-κB (toll-like receptor 2/myeloid differentiation primary response 88/ nuclear factor kappa B) signaling pathway [84][129]. This in turn encourages lung PMN formation through recruitment of monocytes, suppression of T cell function, and enhanced vascular permeability. By reducing the release of LC3+ EVs from these cells or neutralizing CCL2, lung metastasis was inhibited, further supporting the role of EVs in developing the PMN [84][129]. Another group also found that CCL2 expression in the lung could be regulated by 4T1 breast cancer EVs containing miR-200b-3p, which binds to PTEN to stimulate the AKT/NF-κB pathway [85][88]. They observed EV uptake by type II alveolar epithelial cells in vivo which resulted in recruitment of myeloid-derived suppressor cells to promote immune suppression in the lung. The number of lung metastases was significantly higher in EV-treated mice compared to those with CCL2 knockdown, again demonstrating the role of EVs in priming the lung microenvironment for metastatic breast tumor cells [85][88]. Collectively, these findings demonstrate how EVs can modify the behaviour of different cells of the immune system to form the tumor immune microenvironment. These alterations come together to support PMN formation and assist metastatic breast tumor cells in successfully evading immune mechanisms in the lung.
Taken together, the studies described above highlight that EVs secreted by primary breast tumor cells can travel through the circulatory system to the lung in order to influence changes in the lung microenvironment via their effect on stromal cells such as endothelial cells, fibroblasts, macrophages, neutrophils, and T cells. In turn, these changes support formation of the PMN prior to the arrival of metastatic breast tumor cells to promote their survival and growth into successful metastases (Figure 1).
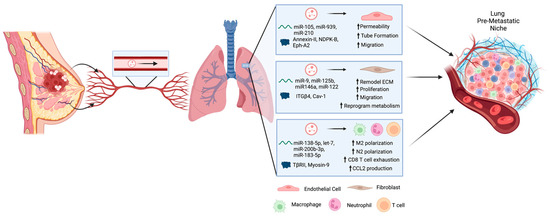
Figure 1. Extracellular vesicles (EVs) are secreted by primary breast tumor cells and travel through the circulatory system to the lung in order to influence changes in the lung microenvironment. These changes are mediated by cargo within the EVs including proteins and RNAs that can be taken up by various stromal cells including endothelial cells, fibroblasts, macrophages, neutrophils, and T cells. Altogether, these changes support formation of the pre-metastatic niche prior to the arrival of metastatic breast tumor cells to promote their survival and growth into successful metastases. Created using BioRender.