Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ho Yu Ng and Version 2 by Sirius Huang.
Despite the declining trend of Helicobacter pylori (H. pylori) prevalence around the globe, ongoing efforts are still needed to optimize current and future regimens in view of the increasing antibiotic resistance. The resistance of H. pylori to different antibiotics is caused by different molecular mechanisms, and advancements in sequencing technology have come a far way in broadening our understanding and in facilitating the testing of antibiotic susceptibility to H. pylori.
- H. pylori
- vonoprazan
- bismuth
1. Introduction
Helicobacter pylori (H. pylori) infects approximately 40% of individuals worldwide [1] and is involved in various gastrointestinal diseases such as chronic gastritis, peptic ulcer disease, upper gastrointestinal bleeding [2], gastric cancer (including gastric adenocarcinoma and gastric MALT lymphoma) [3][4][5][3,4,5] and extraintestinal manifestations [6]. All patients infected with H. pylori should be treated regardless of clinical manifestations [4]. Eradication of H. pylori can restore the normal gastric mucosa [7] and was shown to effectively reduce the development and recurrence of peptic ulcers [8][9][8,9], as well as the incidence of gastric cancer [10][11][10,11].
Various treatment regimens for H. pylori have been developed and their use vary across different geographical regions largely based on local antibiotic resistance patterns. Currently, an acceptable H. pylori treatment regimen is defined as one that achieves at least a 90% cure rate [12], though it has been suggested that an optimized regimen should reliably achieve ≥95% cure rates [13]. Treatment regimens currently recommended by various international guidelines are of empirical nature [4][14][15][16][4,14,15,16]. In light of a substantial decline in efficacy to levels below 80–85% [12][17][12,17], triple therapy consisting of a proton pump inhibitor (PPI), clarithromycin, and amoxicillin or metronidazole is usually not recommended as an empirical first-line therapy for H. pylori infection, except in areas with a known clarithromycin resistance rate of <15% [4][14][15][4,14,15]. Instead, bismuth-based quadruple therapy, typically consisting of bismuth, PPI, metronidazole and tetracycline, is increasingly recommended as its replacement for first-line treatment [4][14][15][4,14,15] and has been shown to achieve over 90% success rate [18][19][18,19]. Bismuth-based quadruple therapy can also be used as a second-line treatment. Another option for second-line treatment is a triple therapy consisting of a PPI, amoxicillin and levofloxacin, but this too faces the problem of decreasing efficacy because of rising resistance to levofloxacin [20]. It is therefore important to deepen our understanding of the mechanisms of antibiotic resistance in H. pylori through the use of various technologies, such as next-generational sequencing (NGS). Antibiotic resistance patterns should be regularly monitored and shared with physicians to guide clinical decisions regarding the antibiotic combinations used for eradication therapy.
2. Antibiotic Resistance in Helicobacter pylori
Primary antibiotic resistance in H. pylori treatment can be defined as resistance in patients who have not started eradication therapy, whereas secondary antibiotic resistance occurs in patients who have previously undergone at least one unsuccessful eradication attempt [21][32]. Globally, primary and secondary resistance rates of clarithromycin, levofloxacin and metronidazole were all over 15% in all World Health Organization (WHO) regions [21][32]. The underlying mechanism behind antibiotic resistance by H. pylori is often due to genetic mutations that may inhibit the intracellular activation of antibiotics or change the drug target site altogether [22][33], and these mutations are mostly encoded chromosomally rather than in extrachromosomal elements [23][24][25][34,35,36]. The specifics of such genetic changes depend on the class of antibiotics (Figure 1) (Table 1). Apart from genetic sequence mutations, there are other possible mechanisms such as efflux pumps through the upregulation or downregulation of hefABC genes, or through biofilm formation, which can obstruct drug penetration [22][33] (Figure 1).
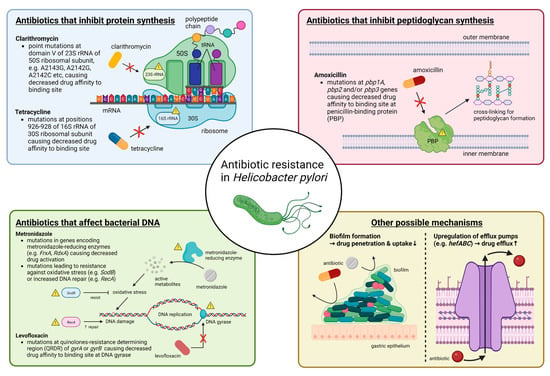
Figure 1. Summary of molecular mechanisms of antibiotic resistance in H. pylori. In general, antibiotic resistance in H. pylori arises because of mutations that decrease drug affinity to its binding site. Examples include mutations in 23S rRNA for clarithromycin, in pbp1A for amoxicillin, in gyrA or gyrB for levofloxacin and in 16S rRNA for tetracycline. Metronidazole is unique in that it is a prodrug and works by producing active metabolites that can damage bacterial DNA after reductive activation. Mutations causing metronidazole resistance have a much more diverse repertoire, and more well-known ones include those in genes encoding for metronidazole-reducing enzymes (e.g., FrxA and RdxA) causing decreased drug activation, as well as those that lead to resistance to oxidative stress caused by metronidazole or increased DNA repair. Other possible mechanisms that may apply to most antibiotics include increased drug efflux caused by upregulation of efflux pump genes (e.g., hefABC) or biofilm formation, which decrease drug penetration. Abbreviations: rRNA, ribosomal RNA; mRNA, messenger RNA; tRNA, transfer RNA; DNA, deoxyribonucleic acid.
Table 1.
Molecular basis of resistance against antibiotics commonly used in
Helicobacter pylori
eradication therapies.
| Genes Involved | Resistance Mechanisms | |
|---|---|---|
| Clarithromycin | Domain V of 23S rRNA (most prevalent: A2143G, A2142G, A2142C) [22][26][27][28][33,37,38,39] | Decrease drug affinity to its binding site |
| infB (translation initiation factor IF-2), Rpl22 (Ribosomal protein L22) [29][30][31][40,41,42] |
Putative effects on 23S rRNA (the drug target) * | |
| Novel gene candidate: fliJ (flagellar export protein): Gln31Arg [32][43] |
Putative effects on bacterial chemotaxis and flagellar motility * | |
| Metronidazole | RdxA (oxygen-insensitive NAD(P)H nitroreductase), FrxA (NAD(P)H flavin nitroreductase), |
Mutations in metronidazole-reducing enzymes coding genes causing decreased drug activation |
| FdxA (ferredoxin), FdxB (ferredoxin-like protein), FldA (flavodoxin) [22][33] |
Mutations in putative metronidazole-reducing enzymes coding genes causing decreased drug activation | |
| SodB (superoxide dismutase), Fur (ferric uptake regulator), MdaB (modulator of drug activity B) [22][33] |
Resistance against oxidative stress brought by metronidazole | |
| RecA [22][33] | Upregulation of RecA causing enhanced DNA repair against damages brought by metronidazole | |
| Ribf (riboflavin biosynthesis protein), Omp11 [22][33] |
Putative association with metronidazole resistance * | |
| hefA (efflux pump) [33][34][44,45] | Increased drug efflux causing below lethal intracellular concentrations | |
| Levofloxacin | gyrA (DNA gyrase subunit A): mainly at codons N87 or D91, gyrB (DNA gyrase subunit B) [22][35][33,46] |
Mutations in quinolones resistance-determining region (QRDR) causing decreased drug affinity to its binding site |
| Novel gene candidates: fliJ (flagellar export protein): Gln31Arg [32][43] cheA (histidine kinase): Ser176Thr/Ala [32][43] |
Putative effects on bacterial chemotaxis and flagellar motility * | |
| Amoxicillin | pbp1A (penicillin-binding protein 1A) [36][37][47,48] pbp2 (penicillin-binding protein 2) [38][49] pbp3 (penicillin-binding protein 3) [38][49] |
Mutations altering penicillin-binding motifs SXXK, SXN and KTG, causing decreased drug affinity to its binding site |
| hopB, hopC (porins), hefC (efflux pump) [39][40][50,51] |
Increased drug efflux causing below lethal intracellular concentrations | |
| Tetracycline | 16S rRNA positions 926–928 [41][42][43][52,53,54] | Decrease drug affinity to its binding site |
| hefABC (efflux pump) [44][55] | Increased drug efflux causing below lethal intracellular concentrations |
* Hypothetical mechanism.
2.1. Resistance to Clarithromycin
Clarithromycin resistance in H. pylori is most commonly due to point mutations in domain V of 23S rRNA, especially A2143G, A2142G and A2142C [22][26][27][28][33,37,38,39], with A2143G causing the lowest eradication rate [45][46][56,57]. Other mutations have also been observed in [47][48][49][58,59,60] and outside of domain V [50][61], as well as in genes outside of 23S rRNA, such as infB and rpl22, which putatively affect 23S rRNA [29][30][31][40,41,42]. Increasing clarithromycin resistance was responsible for the failure of clarithromycin-containing eradication therapies [4][20][51][4,20,62]. The primary clarithromycin resistance rate was below 15% only in the WHO regions of the Americas and Southeast Asia, and when secondary resistance was considered, all WHO regions had >15% resistance rate [21][32], which has exceeded the suggested threshold at which clarithromycin triple therapy could be used as an empirical first-line treatment [52][63]. Studies found that primary clarithromycin resistance rates were 17–27% in Asia [53][54][64,65], 25% in treatment-naïve patients in Europe [55][66], 17.6–31.5% in the US [56][57][67,68] and 29.2% in Africa [58][69].
Of note, resistance caused by prior usage of clarithromycin or other macrolides for other diseases in patients who had never taken clarithromycin-based eradication therapy may be considered as secondary resistance [59][60][70,71]. Information on prior macrolide use, though difficult to obtain in real practice [61][72], should be collected whenever possible as cross resistance can occur within the same family of antibiotics [20]. Studies showed that macrolide exposure even in the past 10 to 14 years still correlated with H. pylori eradication failure [60][61][62][63][64][71,72,73,74,75] and was an independent risk factor for H. pylori clarithromycin resistance [60][71]. One such study found that those who previously took macrolides for >2 weeks had a significantly higher failure rate of eradication than those who took macrolides for ≤2 weeks (44.8% vs. 29.3%, p = 0.047), suggesting a duration effect of prior macrolide use on the success rate of eradication therapy [62][73].
2.2. Resistance to Metronidazole
The mutational changes involved in metronidazole resistance have a much more diverse repertoire and the genotype–phenotype correlation is often much more complex [4][22][4,33]. More well-known ones included RdxA and FrxA mutations, which were metronidazole-reducing enzymes and resulted in reduced drug activation [22][33]. Other putative mutations might result in increased efficiency of deoxyribonucleic acid (DNA) repair or oxidative stress response [22][33] and upregulated efflux [33][34][44,45]. It is possible that metronidazole resistance is caused by the cumulative effects of multiple pathways rather than a single mechanism, though until now, there has been no study that comprehensively investigated these mechanisms in the same clinical isolates [22][33].
The metronidazole resistance rate is generally much higher than that of clarithromycin, in part because of its widespread use to treat parasitic infections, urinary tract infections and gastrointestinal infections by anaerobes [65][76]. The primary and secondary resistance rates of metronidazole were over 15% in all WHO regions, with the highest rates of 56% and 65%, respectively, observed in the eastern Mediterranean region [21][32]. An increasing trend of metronidazole resistance has been observed in most of the WHO regions [21][32]. Studies have found that primary metronidazole resistance was 44% in the Asia-Pacific region [53][64], 30–40% in Europe [55][66][66,77], 42–43% in the US [56][57][67,68] and 48.7% in Africa [67][78]. A notable issue was the dual resistance to clarithromycin and metronidazole, which had a prevalence rate of around 8–15% in Europe [21][55][66][32,66,77], 6–11% in Asia [21][32] and 3–11% in the Americas [21][56][57][32,67,68]. Such dual resistance greatly reduces the efficacy of non-bismuth quadruple therapy [68][69][79,80]. In contrast to clarithromycin, in vitro metronidazole resistance can be overcome by increasing the dose, frequency and duration of therapy [52][70][71][72][63,81,82,83].
2.3. Resistance to Levofloxacin
As H. pylori does not naturally possess the genes for topoisomerase IV, levofloxacin resistance in H. pylori mainly arises from point mutations in the quinolone resistance-determining region (QRDR) of the gyrA or, to a lesser extent, gyrB gene, which code for DNA gyrase subunits A and B, respectively [22][35][33,46]. Mutations at gyrA mainly occur at codons N87 or D91 [73][74][84,85], while E463 mutation has been observed at gyrB [75][76][86,87].
Primary resistance against levofloxacin exceeded 15% in all WHO regions except Europe, while secondary resistance exceeded 15% in all regions with a significant rising trend observed in the western Pacific WHO region [21][32]. Other studies found that the primary levofloxacin resistance rates were 18% in the Asia-Pacific region [53][64], 15–20% in Europe [55][66][66,77], 37.6% in the US [57][68] and 17.4% in Africa [58][69]. Multiple resistance, commonly as dual or triple resistance to clarithromycin and/or metronidazole, is also an area of concern, with rates up to 6–10% in Europe [55][66][66,77].
2.4. Resistance to Amoxicillin
Resistance to amoxicillin in H. pylori are mainly caused by mutations in the pbp1A gene [36][37][47,48] that altered the penicillin-binding motifs SXXK, SXN and KTG, hence reducing the affinity of amoxicillin to PBP [22][26][33,37]. Enhancing effects can be contributed to by mutations in pbp2 and pbp3, and if mutations in these three genes occur simultaneously, resistance could increase by over 200-fold [38][49]. Mutations of porin genes (e.g., hopB and hopC) or efflux pump-coding genes (e.g., hefC) can contribute to amoxicillin resistance in H. pylori as well [39][40][50,51].
Amoxicillin resistance in H. pylori is generally lower than other antibiotics. Primary and secondary amoxicillin resistance rates were previously found to be below 15% in all WHO regions [21][32]. Low resistance was generally found in more-developed regions, such as 6.4% in the US [56][67], 3% in the Asia-Pacific region [53][64] and 0.2% in Europe [66][77]. In Africa, the resistance rate could be as high as 72.6% [58][69], likely caused by the over abuse of amoxicillin because of low cost [77][88].
2.5. Resistance to Tetracycline
Tetracycline resistance in H. pylori is mainly caused by mutations at positions 926–928 of the 16S rRNA [41][42][43][52,53,54] that can lead to reduced drug affinity as they are located at the primary binding site [78][89]. Among these mutations, triple base-pair mutations (e.g., AGA → TTC) conferred a higher level of tetracycline resistance than single or double base-pair mutations [79][90]. Other possible mechanisms included efflux pump mechanisms that might be mediated by HefABC [44][55] or by proton motive force (PMF)-dependent mechanisms [80][91], which might explain resistance in H. pylori strains without 16S rRNA mutations.
Similar to amoxicillin, resistance against tetracycline is generally low around the world, with primary and secondary resistance below 10% in all WHO regions [21][32]. The overall resistance rate of tetracycline was 4% in the Asia-Pacific region [53][64], below 1% in Europe [55][66][66,77] and below 3% in the US [56][57][67,68]. In Africa, however, misuse of tetracyclines caused the resistance rate to be much higher at 49.8% [58][69], which might cause reduced efficacy of bismuth quadruple therapy locally [52][63].