In dilated cardiomyopathy (DCM), where the heart muscle becomes stretched and thin, heart failure (HF) occurs, and the cardiomyocytes suffer from an energetic inefficiency caused by an abnormal cardiac metabolism. Although underappreciated as a potential therapeutic target, the optimal metabolic milieu of a failing heart is still largely unknown and subject to debate. Because glucose naturally has a lower P/O ratio (the ATP yield per oxygen atom), the previous studies using this strategy to increase glucose oxidation have produced some intriguing findings. In reality, the vast majority of small-scale pilot trials using trimetazidine, ranolazine, perhexiline, and etomoxir have demonstrated enhanced left ventricular (LV) function and, in some circumstances, myocardial energetics in chronic ischemic and non-ischemic HF with a reduced ejection fraction (EF).
- dilated cardiomyopathy
- heart failure
- metabolic therapy
- myocardial energetics
- SGLT2 inhibitors
1. The Purpose of Metabolic Therapy in Dilated Cardiomyopathy
2.1. Betablockers
2.2. RAAS Inhibitors
2.3. Angiotensin Receptor Neprilysin Inhibitors (ARNI)
2.4. Mineralocorticoid Receptor Antagonists (MRAs)
2.5. Loop Diuretics
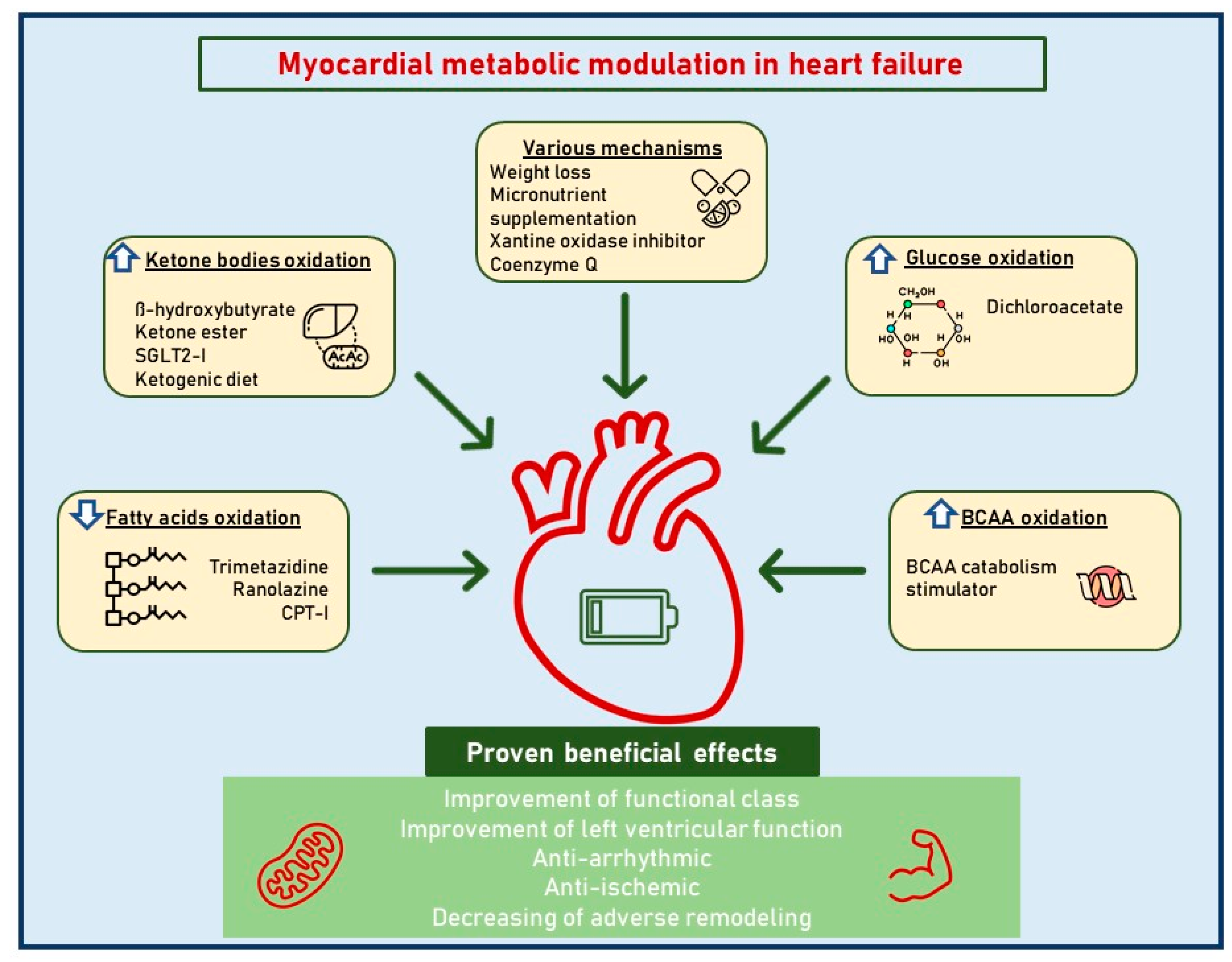
3. Direct Cardiac Metabolism Modulators
4. The Ketone Bodies Hypothesis
As outlined above, targeting cardiac metabolism by decreasing fatty acid oxidation and promoting glucose oxidation appears to be an interesting approach to the treatment of chronic HF. Several drugs have been investigated in small-scale studies, but large clinical trials are needed to confirm the efficacy of these agents as a part of chronic HF treatment. More recently, a new interest in ketone body metabolism has arisen as their modulation may be of potential benefit to HF patients. Under normal conditions, ketones represent a minimal part of all substrates utilized by the myocardium for energy production. These compounds, however, become critical during periods of stress and fasting since their utilization allows the preservation of glycogen stores. The myocardium is the highest ketone body consumer per unit mass. Ketone body oxidation is also more efficient than fatty acid oxidation in terms of ATP synthesis per molecule of oxygen used [65][66]. In addition, ketone body metabolism exerts anti-oxidant effects since it oxidizes mitochondrial co-enzyme Q and reduces cytosolic [NADP+]/[NADPH+], thereby decreasing free radical production [66]. The resourcefulness of the heart in using ketone bodies in order to satisfy its ATP requirements serves as a tool to spare glucose. However, it remains rather unclear whether their employment is compensative to balance out the negative effects of the failing heart adaptive/maladaptive substrate utilization. In hypertrophied and early-stage failing rat hearts, a reduced capacity to oxidize fatty acids and a shift to ketone oxidation as an alternative metabolic fuel have been observed [67]. Similar data have been found in failing human hearts: patients with reduced LV EF nearly tripled their consumptions of ketones as metabolic substrates compared to patients with preserved EF [27]. A case control study involving patients with chronic dilated non-ischemic cardiomyopathy showed increased amounts of beta-hydroxybutyryl CoA and decreased amounts of myocardial beta-hydroxybutyrate in myocardial tissue, suggesting an increased ketone body metabolism in this setting [68]. Additional studies have shown that circulating ketone bodies in subjects with chronic HF increase proportionally to the intensity of their symptoms, the level of congestion in the venous circulatory system, and the magnitude of neurohormonal and cytokine involvement, as well as the increasing deterioration of left ventricular function [69][70]. In this context, more ketones are produced through hepatic ketogenesis and become a fundamental substrate for energy production in cardiomyocytes [71]. These results clearly indicate that chronic HF determines a ketosis-prone state [69]. Indeed, exhaled acetone levels have been shown to be able to identify HF patients with a predictive value which is somewhat similar to that of brain natriuretic peptide (BNP); moreover, this predictive value is proportional to the NYHA class [70]. It is also known that exhaled breath acetone is increased in HF patients with reduced EF and is associated with higher mortality or heart transplantation [72]. Interestingly, higher serum levels of beta-hydroxybutyrate seem to relate to disease progression and adverse prognosis in arrhythmogenic cardiomyopathy patients, supporting the hypothesis that an enhanced ketone body metabolism may be a standard myocardium response to injuries [73]. According to another study, the cardiomyocytes’ specific loss of succinyl-CoA:3-oxoacid CoA transferase, which is involved in ketone body oxidation, is associated with significantly increased left ventricular volume and a decreased ejection fraction as a response to pressure overload [74]. Overall, these studies confirm the fundamental role of this metabolic pathway, showing that impaired ketone body oxidation may be associated with worsened heart remodeling following pressure overload. In this context, the concept of the therapeutic modulation of ketone metabolism as a potential new target in HF treatment is emerging [74][75][76][77][78].5. Sodium-Glucose Cotransporter-2 Inhibitors (SGLT2i)
There is a bidirectional link between diabetes mellitus (DM) and HF. Longstanding diabetes causes changes in myocardial metabolism, abnormal calcium signaling, and inflammatory pathways, resulting in structural and functional changes in the myocardium and leading to the development of diabetic cardiomyopathy and the progression of HF [79][80]. Conversely, HF patients without DM are at an increased risk of developing glycemic abnormalities [79]. The shared underlying risk factors and the overlap of the pathophysiological mechanisms play a critical role in the frequent coexistence of DM and HF. As with HF, there is also a strong link between diabetes, coronary artery disease, hypertension, and renal disease. During the last decade, cardiovascular outcome trials have investigated several classes of new glucose-lowering agents, including SGLT2i, which, apart from showing evidence of cardiovascular safety, have also been shown to exert beneficial effects on the cardiovascular outcome [81][82]. Most studies have shown the independence of cardiovascular outcome from glycemic control, indicating mechanisms of action other than those usually postulated to explain the cardiovascular benefits of glucose-lowering therapies [83][84][85][86][87][88]. In fact, the significant beneficial clinical effects observed with SGLT2i use cannot be explained by one single mechanism.References
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276.
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858.
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414.
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. J. Intern. Med. 2019, 286, 362–372.
- Lüscher, T.F. Heart failure: The cardiovascular epidemic of the 21st century. Eur. Heart J. 2015, 36, 395–397.
- Bleumink, G.S.; Knetsch, A.M.; Sturkenboom, M.C.; Straus, S.M.; Hofman, A.; Deckers, J.W.; Witteman, J.C.; Stricker, B.H. Quantifying the heart failure epidemic: Prevalence, incidence rate, lifetime risk and prognosis of heart failure The Rotterdam Study. Eur. Heart J. 2004, 25, 1614–1619.
- van Riet, E.E.; Hoes, A.W.; Wagenaar, K.P.; Limburg, A.; Landman, M.A.; Rutten, F.H. Epidemiology of heart failure: The prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur. J. Heart Fail. 2016, 18, 242–252.
- Urbich, M.; Globe, G.; Pantiri, K.; Heisen, M.; Bennison, C.; Wirtz, H.S.; Di Tanna, G.L. A Systematic Review of Medical Costs Associated with Heart Failure in the USA (2014–2020). Pharmacoeconomics 2020, 38, 1219–1236.
- Girerd, N.; Von Hunolstein, J.J.; Pellicori, P.; Bayés-Genís, A.; Jaarsma, T.; Lund, L.H.; Bilbault, P.; Boivin, J.M.; Chouihed, T.; Costa, J.; et al. Therapeutic inertia in the pharmacological management of heart failure with reduced ejection fraction. ESC Heart Fail. 2022, 9, 2063–2069.
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258.
- Ingwall, J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009, 81, 412–419.
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726.
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032.
- Jones, N.R.; Roalfe, A.K.; Adoki, I.; Hobbs, F.D.R.; Taylor, C.J. Survival of patients with chronic heart failure in the community: A systematic review and meta-analysis. Eur. J. Heart Fail. 2019, 21, 1306–1325.
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513.
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129.
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68.
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151.
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726.
- Tian, R.; Colucci, W.S.; Arany, Z.; Bachschmid, M.M.; Ballinger, S.W.; Boudina, S.; Bruce, J.E.; Busija, D.W.; Dikalov, S.; Dorn, G.W., II; et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System: Path to a Cure in Heart Failure—A Report. from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation 2019, 140, 1205–1216.
- Allard, M.F.; Schönekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267 Pt 2, H742–H750.
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724.
- Despa, S.; Bers, D.M. Na⁺ transport in the normal and failing heart—Remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10.
- Barger, P.M.; Kelly, D.P. Fatty acid utilization in the hypertrophied and failing heart: Molecular regulatory mechanisms. Am. J. Med. Sci. 1999, 318, 36–42.
- Funada, J.; Betts, T.R.; Hodson, L.; Humphreys, S.M.; Timperley, J.; Frayn, K.N.; Karpe, F. Substrate utilization by the failing human heart by direct quantification using arterio-venous blood sampling. PLoS ONE 2009, 4, e7533.
- Voros, G.; Ector, J.; Garweg, C.; Droogne, W.; Van Cleemput, J.; Peersman, N.; Vermeersch, P.; Janssens, S. Increased Cardiac Uptake of Ketone Bodies and Free Fatty Acids in Human Heart Failure and Hypertrophic Left Ventricular Remodeling. Circ. Heart Fail. 2018, 11, e004953.
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368.
- Chen, L.; Song, J.; Hu, S. Metabolic remodeling of substrate utilization during heart failure progression. Heart Fail. Rev. 2019, 24, 143–154.
- Larsen, T.S.; Jansen, K.M. Impact of Obesity-Related Inflammation on Cardiac Metabolism and Function. J. Lipid Atheroscler. 2021, 10, 8–23.
- Raghow, R. An ‘Omics’ Perspective on Cardiomyopathies and Heart Failure. Trends Mol. Med. 2016, 22, 813–827.
- Ampong, I. Metabolic and Metabolomics Insights into Dilated Cardiomyopathy. Ann. Nutr. Metab. 2022, 78, 147–155.
- Flam, E.; Jang, C.; Murashige, D.; Yang, Y.; Morley, M.P.; Jung, S.; Kantner, D.S.; Pepper, H.; Bedi, K.C., Jr.; Brandimarto, J.; et al. Integrated landscape of cardiac metabolism in end-stage human nonischemic dilated cardiomyopathy. Nat. Cardiovasc. Res. 2022, 1, 817–829.
- Thompson, D.S.; Naqvi, N.; Juul, S.M.; Coltart, D.J.; Jenkins, B.S.; Webb-Peploe, M.M. Haemodynamic and metabolic effects of atenolol in patients with angina pectoris. Br. Heart J. 1980, 43, 668–679.
- Day, J.L. The metabolic consequences of adrenergic blockade: A review. Metabolism 1975, 24, 987–996.
- Lech, J.J.; Jesmok, G.J.; Calvert, D.N. Effects of drugs and hormones on lipolysis in heart. Fed. Proc. 1977, 36, 2000–2008.
- Simonsen, S.; Ihlen, H.; Kjekshus, J.K. Haemodynamic and metabolic effects of timolol (Blocadren) on ischaemic myocardium. Acta Med. Scand. 1983, 213, 393–398.
- Nielsen, T.T.; Bagger, J.P.; Thomassen, A. Improved myocardial lactate extraction after propranolol in coronary artery disease: Effected by peripheral glutamate and free fatty acid metabolism. Br. Heart J. 1986, 55, 140–147.
- Pisarenko, O.I.; Solomatina, E.S.; Studneva, I.M.; Ivanov, V.E.; Kapelko, V.I.; Smirnov, V.N. Protective effect of glutamic acid on cardiac function and metabolism during cardioplegia and reperfusion. Basic. Res. Cardiol. 1983, 78, 534–543.
- Spoladore, R.; Fragasso, G.; Perseghin, G.; De Cobelli, F.; Esposito, A.; Maranta, F.; Calori, G.; Locatelli, M.; Lattuada, G.; Scifo, P.; et al. Beneficial effects of beta-blockers on left ventricular function and cellular energy reserve in patients with heart failure. Fundam. Clin. Pharmacol. 2013, 27, 455–464.
- Fragasso, G.; Salerno, A.; Margonato, A. Heart rate reduction is probably not the main beneficial mechanism by which beta blockade improves outcome in patients with systolic chronic heart failure. Am. J. Cardiol. 2008, 102, 506–507.
- Al-Hesayen, A.; Azevedo, E.R.; Floras, J.S.; Hollingshead, S.; Lopaschuk, G.D.; Parker, J.D. Selective versus nonselective beta-adrenergic receptor blockade in chronic heart failure: Differential effects on myocardial energy substrate utilization. Eur. J. Heart Fail. 2005, 7, 618–623.
- Podbregar, M.; Voga, G. Effect of selective and nonselective beta-blockers on resting energy production rate and total body substrate utilization in chronic heart failure. J. Card. Fail. 2002, 8, 369–378.
- Reneland, R.; Alvarez, E.; Andersson, P.E.; Haenni, A.; Byberg, L.; Lithell, H. Induction of insulin resistance by beta-blockade but not ACE-inhibition: Long-term treatment with atenolol or trandolapril. J. Hum. Hypertens. 2000, 14, 175–180.
- Paolisso, G.; De Riu, S.; Marrazzo, G.; Verza, M.; Varricchio, M.; D’Onofrio, F. Insulin resistance and hyperinsulinemia in patients with chronic congestive heart failure. Metabolism 1991, 40, 972–977.
- Ferrua, S.; Bobbio, M.; Catalano, E.; Grassi, G.; Massobrio, N.; Pinach, S.; Rossi, C.; Veglio, M.; Trevi, G.P. Does carvedilol impair insulin sensitivity in heart failure patients without diabetes? J. Card. Fail. 2005, 11, 590–594.
- Poole-Wilson, P.A.; Swedberg, K.; Cleland, J.G.; Di Lenarda, A.; Hanrath, P.; Komajda, M.; Lubsen, J.; Lutiger, B.; Metra, M.; Remme, W.J. Carvedilol or Metoprolol European Trial Investigators. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol or Metoprolol European Trial (COMET): Randomised controlled trial. Lancet 2003, 362, 7–13.
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: A metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail. 2012, 5, 493–503.
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., 2nd; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846.
- Pellieux, C.; Aasum, E.; Larsen, T.S.; Montessuit, C.; Papageorgiou, I.; Pedrazzini, T.; Lerch, R. Overexpression of angiotensinogen in the myocardium induces downregulation of the fatty acid oxidation pathway. J. Mol. Cell. Cardiol. 2006, 41, 459–466.
- Mori, J.; Alrob, O.A.; Wagg, C.S.; Harris, R.A.; Lopaschuk, G.D.; Oudit., G.Y. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: A critical role of PDK4. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1103–H1113.
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090.
- Vermes, E.; Ducharme, A.; Bourassa, M.G.; Lessard, M.; White, M.; Tardif, J.C. Enalapril reduces the incidence of diabetes in patients with chronic heart failure: Insight from the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 2003, 107, 1291–1296.
- Yusuf, S.; Ostergren, J.B.; Gerstein, H.C.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Olofsson, B.; Probstfield, J.; McMurray, J.V. Candesartan in Heart Failure-Assessment of Reduction in Mortality and Morbidity Program Investigators. Effects of candesartan on the development of a new diagnosis of diabetes mellitus in patients with heart failure. Circulation 2005, 112, 48–53.
- Nuzzi, V.; Raafs, A.; Manca, P.; Henkens, M.T.; Gregorio, C.; Boscutti, A.; Verdonschot, J.; Hazebroek, M.; Knackstedt, C.; Merlo, M.; et al. Left Atrial Reverse Remodeling in Dilated Cardiomyopathy. J. Am. Soc. Echocardiogr. 2023, 36, 154–162.
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004.
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653.
- Seferovic, J.P.; Claggett, B.; Seidelmann, S.B.; Seely, E.W.; Packer, M.; Zile, M.R.; Rouleau, J.L.; Swedberg, K.; Lefkowitz, M.; Shi, V.C.; et al. Effect of sacubitril/valsartan versus enalapril on glycaemic control in patients with heart failure and diabetes: A post-hoc analysis from the PARADIGM-HF trial. Lancet Diabetes Endocrinol. 2017, 5, 333–340.
- Armentaro, G.; D’Arrigo, G.; Miceli, S.; Cassano, V.; Perticone, M.; Maio, R.; Marra, A.M.; Arturi, F.; Cittadini, A.; Tripepi, G.; et al. Long Term Metabolic Effects of Sacubitril/Valsartan in Non-Diabetic and Diabetic Patients with Heart Failure Reduced Ejection Fraction: A Real Life Study. Front. Physiol. 2022, 13, 897109.
- Falch, D.K.; Schreiner, A. The effect of spironolactone on lipid, glucose and uric acid levels in blood during long-term administration to hypertensives. Acta Med. Scand. 1983, 213, 27–30.
- Yamaji, M.; Tsutamoto, T.; Kawahara, C.; Nishiyama, K.; Yamamoto, T.; Fujii, M.; Horie, M. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A₁(c) levels in patients with chronic heart failure. Am. Heart J. 2010, 160, 915–921.
- Korol, S.; Mottet, F.; Perreault, S.; Baker, W.L.; White, M.; de Denus, S. A systematic review and meta-analysis of the impact of mineralocorticoid receptor antagonists on glucose homeostasis. Medicine 2017, 96, e8719.
- Preiss, D.; van Veldhuisen, D.J.; Sattar, N.; Krum, H.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B.; Zannad, F.; et al. Eplerenone and new-onset diabetes in patients with mild heart failure: Results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Eur. J. Heart Fail. 2012, 14, 909–915.
- Fragasso, G.; Margonato, A.; Spoladore, R.; Lopashuck, G.D. Metabolic effects of cardiovascular drugs. Trends Cardiovasc. Med. 2019, 29, 176–187.
- Fragasso, G. Deranged Cardiac Metabolism and the Pathogenesis of Heart Failure. Card. Fail. Rev. 2016, 2, 8–13.
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell. Metab. 2017, 25, 262–284.
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fatty Acids. 2004, 70, 309–319.
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705.
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716.
- Lommi, J.; Kupari, M.; Koskinen, P.; Näveri, H.; Leinonen, H.; Pulkki, K.; Härkönen, M. Blood ketone bodies in congestive heart failure. J. Am. Coll. Cardiol. 1996, 28, 665–672.
- Marcondes-Braga, F.G.; Gutz, I.G.R.; Batista, G.L.; Saldiva, P.H.N.; Ayub-Ferreira, S.M.; Issa, V.S.; Mangini, S.; Bocchi, E.A.; Bacal, F. Exhaled acetone as a new biomaker of heart failure severity. Chest 2012, 142, 457–466.
- Schugar, R.C.; Moll, A.R.; André d’Avignon, D.; Weinheimer, C.J.; Kovacs, A.; Crawford, P.A. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol. Metab. 2014, 3, 754–769.
- Marcondes-Braga, F.G.; Batista, G.L.; Gutz, I.G.; Saldiva, P.H.; Mangini, S.; Issa, V.S.; Ayub-Ferreira, S.M.; Bocchi, E.A.; Pereira, A.C.; Bacal, F. Impact of Exhaled Breath Acetone in the Prognosis of Patients with Heart Failure with Reduced Ejection Fraction (HFrEF). One Year of Clinical Follow-up. PLoS ONE 2016, 11, e0168790.
- Song, J.P.; Chen, L.; Chen, X.; Ren, J.; Zhang, N.N.; Tirasawasdichai, T.; Hu, Z.L.; Hua, W.; Hu, Y.R.; Tang, H.R.; et al. Elevated plasma β-hydroxybutyrate predicts adverse outcomes and disease progression in patients with arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2020, 12, eaay8329.
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019, 4, e124079.
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment with the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141.
- Yurista, S.R.; Silljé, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.M.; Pavez Giani, M.G.; Hillebrands, J.L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873.
- Yurista, S.R.; Chong, C.R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic Potential of Ketone Bodies for Patients with Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 1660–1669.
- Monzo, L.; Sedlacek, K.; Hromanikova, K.; Tomanova, L.; Borlaug, B.A.; Jabor, A.; Kautzner, J.; Melenovsky, V. Myocardial ketone body utilization in patients with heart failure: The impact of oral ketone ester. Metabolism 2021, 115, 154452.
- Oktay, A.A.; Aktürk, H.K.; Paul, T.K.; O’Keefe, J.H.; Ventura, H.O.; Koch, C.A.; Lavie, C.J.; Feingold, K.R.; Anawalt, B.; Boyce, A.; et al. Diabetes, Cardiomyopathy, and Heart Failure. In Endotext ; MDText.com, Inc.: South Dartmouth, MA, USA, 2000.
- Li, N.; Zhou, H. SGLT2 Inhibitors: A Novel Player in the Treatment and Prevention of Diabetic Cardiomyopathy. Drug. Des. Dev. Ther. 2020, 14, 4775–4788.
- Ahmad, Y.; Madhavan, M.V.; Stone, G.W.; Francis, D.P.; Makkar, R.; Bhatt, D.L.; Howard, J.P. Sodium-glucose cotransporter 2 inhibitors in patients with heart failure: A systematic review and meta-analysis of randomized trials. Eur. Heart J. Qual. Care Clin. Outcomes 2022, 8, 383–390.
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829.
- Giugliano, D.; Longo, M.; Scappaticcio, L.; Bellastella, G.; Maiorino, M.I.; Esposito, K. SGLT-2 inhibitors and cardiorenal outcomes in patients with or without type 2 diabetes: A meta-analysis of 11 CVOTs. Cardiovasc. Diabetol. 2021, 20, 236.
- Inzucchi, S.E.; Kosiborod, M.; Fitchett, D.; Wanner, C.; Hehnke, U.; Kaspers, S.; George, J.T.; Zinman, B. Improvement in Cardiovascular Outcomes with Empagliflozin Is Independent of Glycemic Control. Circulation 2018, 138, 1904–1907.
- Petrie, M.C.; Verma, S.; Docherty, K.F.; Inzucchi, S.E.; Anand, I.; Belohlávek, J.; Böhm, M.; Chiang, C.E.; Chopra, V.K.; de Boer, R.A.; et al. Effect of Dapagliflozin on Worsening Heart Failure and Cardiovascular Death in Patients with Heart Failure with and Without Diabetes. JAMA 2020, 323, 1353–1368.
- Williams, D.M.; Evans, M. Are SGLT-2 Inhibitors the Future of Heart Failure Treatment? The EMPEROR-Preserved and EMPEROR-Reduced Trials. Diabetes Ther. 2020, 11, 1925–1934.
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128.
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of Sodium-Glucose Cotransporter 2 Inhibitors for the Treatment of Patients with Heart Failure: Proposal of a Novel Mechanism of Action. JAMA Cardiol. 2017, 2, 1025–1029.