Vascular smooth muscle cells (VSMCs) are the predominant cell type in the medial layer of the aorta, which plays a critical role in the maintenance of aortic wall integrity. VSMCs have been suggested to have contractile and synthetic phenotypes and undergo phenotypic switching to contribute to the deteriorating aortic wall structure. Recently, tThe unprecedented heterogeneity and diversity of VSMCs and their complex relationship to aortic aneurysms (AAs) have been revealed by high-resolution research methods, such as lineage tracing and single-cell RNA sequencing. The aortic wall consists of VSMCs from different embryonic origins that respond unevenly to genetic defects that directly or indirectly regulate VSMC contractile phenotype. This difference predisposes to hereditary AAs in the aortic root and ascending aorta.
- abdominal aortic aneurysm
- thoracic aortic aneurysm
- VSMC
1. Introduction
2. VSMC Heterogeneity in Normal Aorta
The unique embryonic context is a source of VSMC heterogeneity. As reviewed by Majesky, fate mapping technology in developing embryos has revealed a mosaic distribution of VSMCs from different precursor sources in the aorta, which includes VSMCs derived from the secondary heart fields (SHF), cardiac neural crest (CNC), somite, and splanchnic mesoderm [19][17]. For instance, VSMCs of the aortic root are mainly of SHF origin, those of the ascending aorta and aortic arch originate mainly from the CNC, and VSMCs composing the descending aorta are derived from mesoderm [20,21,22][18][19][20]. Even the same aortic segment can consist of VSMCs of different embryonic origins. SHF-derived VSMCs were previously thought to be restricted in the aortic root, forming a suture with CNC-derived VSMCs at the transition from the aortic root and the ascending aorta. However, lineage tracing shows that SHF-derived VSMCs extend distally to the innominate artery orifice. The ascending aorta actually contains both SHF-derived and CNC-derived VSMCs, with the SHF-derived VSMCs wrapping around the outside of the CNC-derived VSMCs in a sleeve-shaped form (Figure 1A) [20,23][18][21].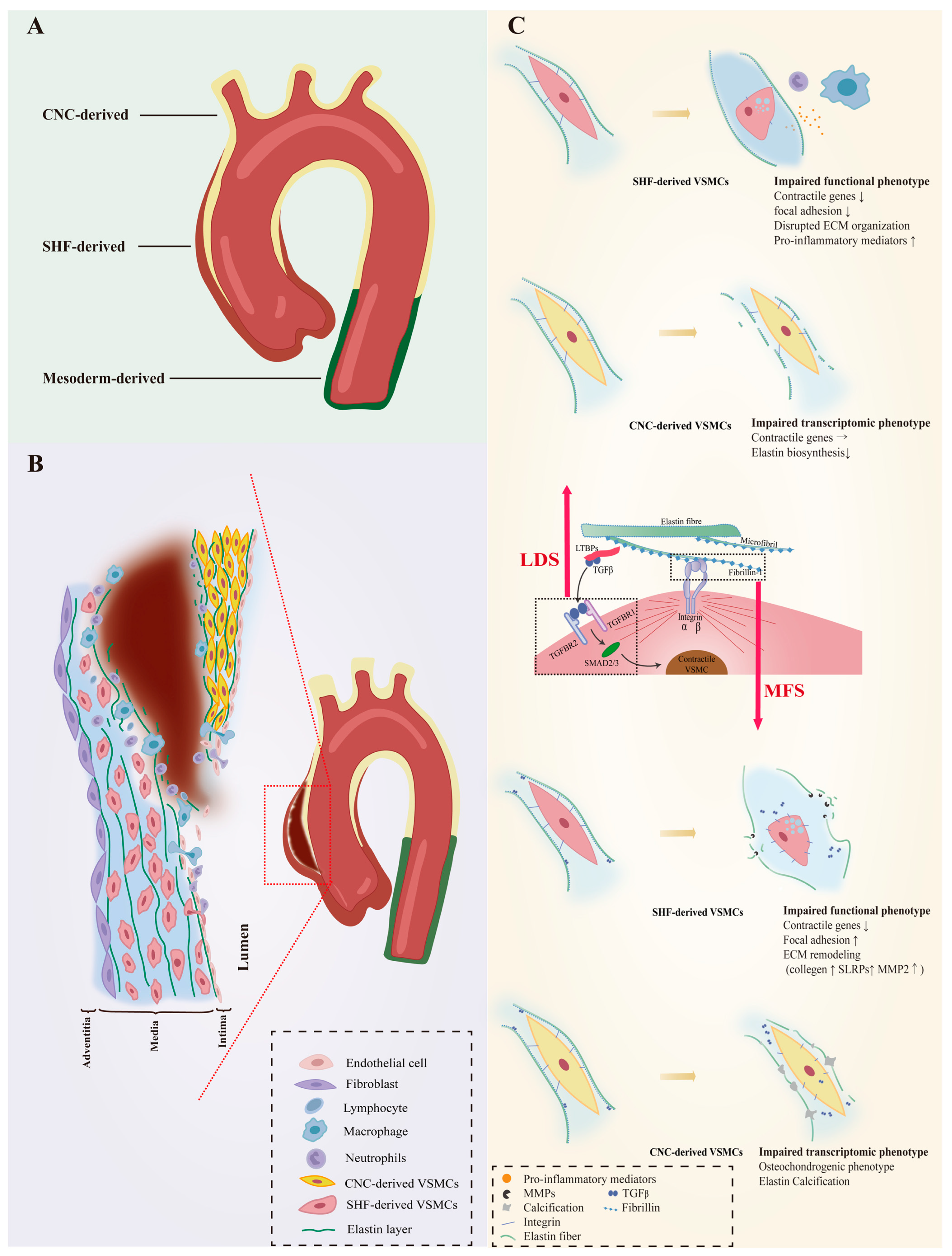
3. VSMC Phenotypic Diversity in Hereditary AAs
The susceptibility to aortopathy varies between aortic segments. The ascending aorta is less prone to atherosclerosis than the abdominal aorta, and even thoracoabdominal aortic graft exchange does not alter atherosclerotic propensity, suggesting that the intrinsic nature of the arterial wall influences vulnerability to arterial disease [42,43,44][34][35][36]. Segment-specific distribution of heterogeneous VSMCs likely contributes significantly to aortic disease characteristics. Further support for this view is the fact that the aortic arch, which is composed of CNC-derived VSMCs, is the segment of the thoracic aorta that is more susceptible to calcification [45,46][37][38]. As VSMCs of different embryonic origins have heterogeneous patterns in response to environmental cues, genetic mutations that can affect intracellular signaling could also have heterogeneous effects on them. Patients with genetic defects in focal adhesion mediators or components of the TGF-β signaling cascade are predisposed to thoracic aortic aneurysms (TAAs) with manifestations of syndromic TAAs such as Marfan syndrome (MFS), Loeys–Dietz syndrome (LDS), or TAAs alone [47,48,49,50,51,52,53,54,55][39][40][41][42][43][44][45][46][47]. Importantly, almost all AAs in LDS and MFS patients occur in the aortic root and ascending arteries, and dissections often arise in the ascending aorta (Figure 1B), suggesting that heterogeneous lineage plasticity of VSMCs determines the pathological characteristics of hereditary aneurysms [56,57][48][49].3.1. VSMC Phenotypic Diversity in LDS
LDS is an aneurysm-predisposing disease caused by defects in the canonical positive regulators of TGF-β, which regulate the differentiation and maturation of VSMCs [58,59][50][51]. Heterozygous loss-of-function mutations in genes encoding multiple components of the TGF-β signaling pathway, including the ligands (TGFB2/3), transforming growth factor-β receptor types I and II (TGFBR1 and TGFBR2) and downstream effectors (SMAD2/3), cause abnormalities in the function of VSMCs (Figure 1C). Approximately 20–25% of patients with LDS have mutations in the TGFBR1 genes [60][52]. The embryonic background of the VSMCs determines the effect of this genetic defect. SHF-derived VSMCs, but not CNC-derived VSMCs, exhibited impaired Smad2/3 activation, increased angiotensin II type 1 receptor (Agtr1a), and TGF-β ligands expression in the LDS model carrying heterozygous Tgfbr1 inactivation [61][53]. A human-induced pluripotent stem cell (hiPSC) model derived from an LDS patient family with the TGFBR1A230T variant also detected disruption of SMAD3 and AKT activation and significantly reduced contractile transcript and protein levels in SHF-derived VSMCs, but not in CNC-derived VSMCs [62][54]. Single-cell transcriptomic data revealed molecular similarities between SHF-derived VSMCs with the TGFBR1A230T variant and SHF-derived VSMCs with a loss-of-function SMAD3 mutation. Additionally, pharmacological activation of the intracellular SMAD2/3 signaling cascade can rescue contractile gene expression and contractile function in TGFBR1 mutant SHF-derived VSMCs [62][54]. Approximately 55–60% of LDS patients carry Tgfbr2 mutations [60][52]. The effect of Tgfbr2-mediated signaling activation on VSMC gene expression depends on the location of VSMCs in the aorta [68][55]. Smooth muscle specific Tgfbr2 deficiency significantly enhances the progression of ascending but not abdominal aortic pathology, including increased intramural hemorrhage, medial thinning, and epicardial thickening, and damage was more pronounced outside of the media layer than inside [69,70][56][57]. Although rigorous experimental evidence such as lineage-stratified sc-RNAseq and lineage tracing is required, it is speculated that VSMCs of different embryonic origins respond heterogeneously to Tgfbr2 deficiency. Whether Tgfbr2 exerts a facilitative or inhibitory effect on the differentiation of neural crest cells (NCCs) into VSMCs remains unclear. TGF-β signaling is involved in NCCs migration and differentiation and germline knockout of Tgfbr2 in VSMCs leads to extensive and lethal cardiovascular malformation in mouse embryos [68,71,72,73][55][58][59][60]. Tgfbr2 deficiency was found to hinder NCC-to-VSMC differentiation, resulting in a deficiency of CNC-derived VSMCs in the aorta [73,74][60][61]. However, recent studies have shown that Tgfbr2 deficiency does not impede the VSMC fate of NCCs and may even lead to premature differentiation of NCCs into VSMCs [68,75,76][55][62][63]. Smad2 is confirmed as an essential regulator in progenitor-specific VSMC development and physiological differences between CNC- and mesoderm-derived VSMCs [72,77,78][59][64][65]. Although in vitro studies found Smad2 functionally intact in Tgfbr2-deficient VSMCs, animal models demonstrated reduced p-Smad2 levels [70,77,79][57][64][66]. Embryonic Smad2 knockdown in NCCs reduces the number of CNC-derived VSMCs and produces a damaged, fragmented elastin lamina in the media of the ascending aorta [70,77,80][57][64][67]. Strikingly, postnatal knockdown of Tgfbr2 in VSMCs, while reducing Tgfb signaling, has little effect on contractile gene expression levels and contractile function in VSMCs, but rapidly induces elastin fragmentation and ascending thoracic aorta disruption (ATAD) [70,76][57][63].3.2. VSMC Phenotypic Diversity in MFS
FBN1 gene mutations predispose Marfan syndrome (MFS) patients to TAA at a young age and premature death from catastrophic aneurysm rupture or dissection [56][48]. The FBN1 gene does not directly regulate the contraction-related phenotype of VSMC, but its coding product fibrillin-1 is an important component of the ECM and acts to mediate the attachment of VSMCs to elastin laminae [89][68]. Fibrillin-1, the encoded product of FBN1, is an important component of the ECM that mediates the attachment of VSMCs to elastin laminae, the loss of which would disrupt mechanotransduction and result in detached VSMCs with altered morphology, reduced contractile signatures, and upregulation of several ECM elements and elastolysis mediators indicating cellular plasticity. (Figure 1C) [90][69]. MFS VSMCs derived from iPSCs showed a lineage-specific protein profile. Compared to CNC-derived VSMCs, SHF-derived VSMCs have a proteome with less VSMC identity (TAGLN), but increased levels of focal adhesion components (integrin αV, fibronectin) and ECM remodeling molecules (collagen type 1, MMP2) [91][70]. As a novel protein marker associated with MFS aneurysms, uPARAP, an endocytic receptor responsible for receptor-mediated internalization of collagen for degradation, was specifically downregulated in SHF-derived VSMCs [91,92][70][71]. Recent work has shown that uPARAP also acts as an endocytic receptor for thrombospondin-1 (TSP-1), which has been reported to promote AAs by increasing aortic inflammation and TGF-β activity [93,94,95,96,97][72][73][74][75][76]. Single-cell transcriptomics provides evidence for lineage-specific VSMC plasticity in MFS aortic aneurysms. Albert and colleagues identified a group of modulated VSMCs (modVSMCs) in aortic aneurysm tissue from Fbn1C1041G/+ mice with a transcriptome similar to that of VSMC-lineage-derived fibromyocytes in atheroma [98][77]. The combination of lineage tracing and scRNA-seq uncovered that SHF-derived VSMCs and CNC-derived VSMCs contribute equally to modVSMCs [99][78].4. VSMC Phenotypic Diversity in Non-Hereditary AAs
Apart from a few that can be explained by causative genetic mutations, most sporadic patients do not have genetic defects and these AAs are called non-hereditary AAs [103,104,105][79][80][81]. The etiology of non-hereditary AAs is still enigmatic and previous studies have shown that risk factors for non-hereditary AAs, such as hypertension, aging, and hyperlipidemia, transform contractile VSMCs into synthetic VSMCs [105,106,107,108,109,110][81][82][83][84][85][86]. However, the definition of synthetic VSMCs is ambiguous and recent studies demonstrate that they are actually conglomerates of several VSMC phenotypes with different functions (Figure 2).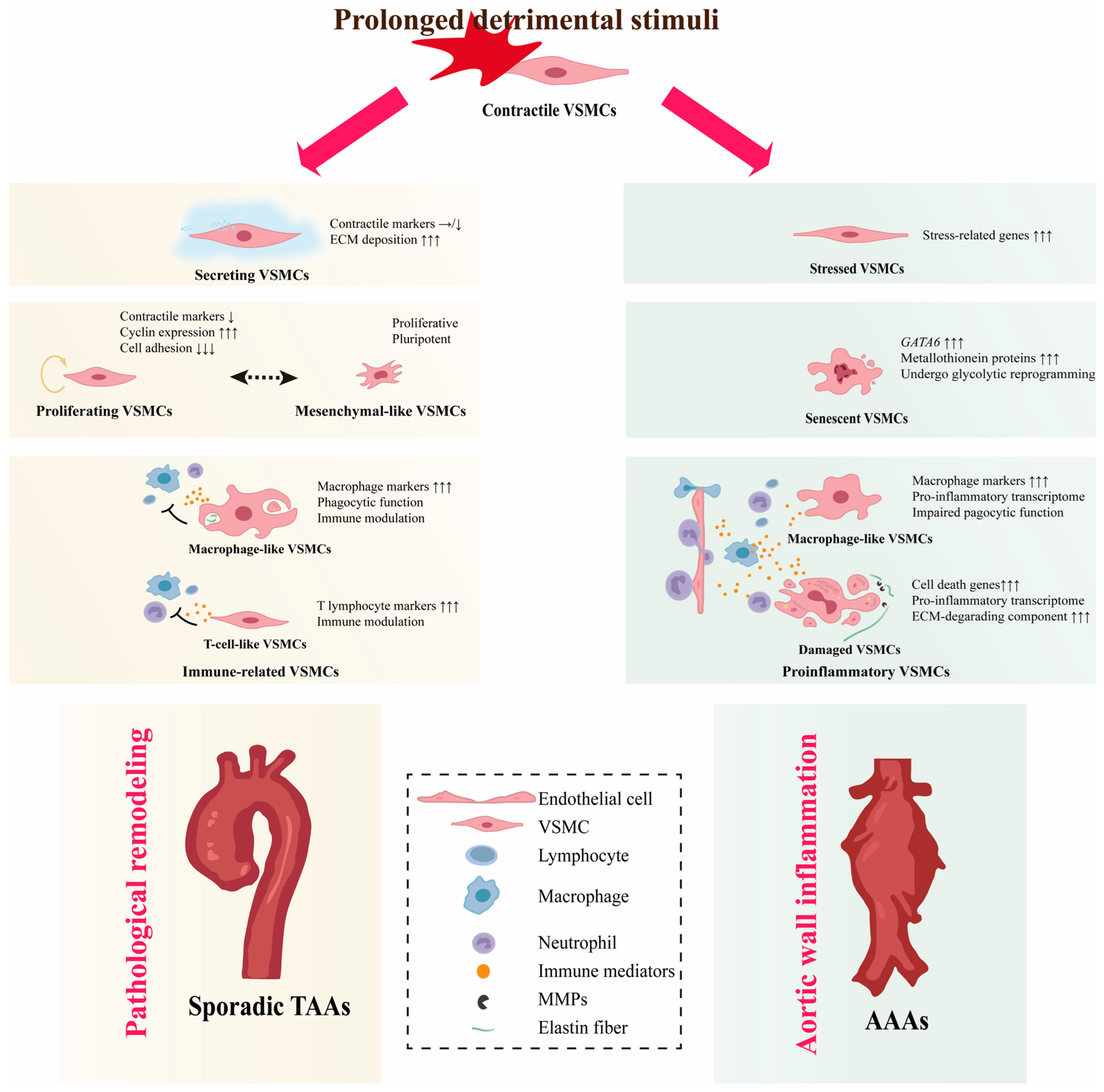
4.1. VSMC Diversity in TAAs
4.1.1. Secreting VSMCs
4.1.2. Proliferating VSMCs
VSMC proliferation is a hallmark of occlusive aortic diseases such as restenosis after angioplasty or stenting [124][94]. Upon injury to the vascular wall, the quiescent, differentiated VSMCs have been reported to be activated to a proliferative phenotype at low frequencies and such oligoclonal contribution of VSMCs is a feature of obstructive arterial disease [125,126,127,128][95][96][97][98]. Fate mapping analyses in TAA models induced by disruption of the TGF-β pathway also revealed oligoclonal expanding of the VSMCs [115,116][99][100]. Enhanced VSMC proliferation was demonstrated in sporadic TAAs [111,112,114][87][88][101]. In human TAA samples, sc-RNAseq identified a subpopulation of VSMCs characterized by high levels of cyclin expression and low levels of cell adhesion, which is highly suggestive of ongoing cell proliferation and was annotated as proliferating VSMCs [111][87]. Despite expressing some canonical synthetic marker genes such as MGP, TPM4, and MYH10, the proliferating VSMCs do not upregulate ECM and inflammatory genes, which is different from synthetic VSMCs [111][87].4.1.3. Immune-Related VSMCs
In the context of atherosclerosis, VSMCs have been shown to have the potential to switch to monocyte/macrophage lineages, implicating them in the modulation of inflammation in the vascular wall [128,129,130,131][98][102][103][104]. Although medial degeneration has been confirmed as the most common pathological manifestation of TAAs, aortitis, and atherosclerosis remain the main histopathological substrates in 1/3 of TAA patients [132,133][105][106]. The prevalence of inflammation-related histologic changes underscores the pivotal role of the inflammatory response in the pathophysiology of TAAs. VSMCs that exhibit similar characteristics of monocyte/macrophage and T-lymphocyte are identified in TAAs and referred to as immune-related VSMCs [111,112,117][87][88][107]. Additionally, an intermediate state, which expresses interferon-induced genes, has the potency to switch to T-cell-like VSMCs or macrophage-like VSMCs, suggesting that VSMCs are activated by inflammation [111][87]. Bioinformatics analysis revealed that immune-related VSMCs exert immunomodulatory functions through rich and strong interactions with immune cells [112][88]. T-cell-like VSMCs recruit immune cells mainly through the CXCL12-CXCR4 pair. Macrophage-like VSMCs in TAAs can communicate with all infiltrating immune cells through PTN and galectin, which are related to the suppressive immune microenvironment in malignancies [134,135,136][108][109][110]. The immunosuppressive function of macrophage-like VSMCs may explain why the inflammatory response in Ang II-induced TAAs is not as strong as in AAAs.4.2. VSMC Diversity in AAAs
4.2.1. Proinflammatory VSMCs
AAA is considered to be a chronic inflammatory disease. Pro-inflammatory VSMCs defined by their function to promote aortic wall inflammation are identified. They have a proinflammatory transcriptome characterized by the expression of proinflammatory cytokines (IL-1ß, IL-6, CCL2, CCL5, and TNFα), chemokines (CXCL10 and ICAM1), and high expression of ECM-degrading components such as MMP2/9. Aortic wall inflammation can damage VSMCs, activate the expression of inflammation-related genes through innate immune pathways, and further promote vascular wall inflammation, forming a vicious cycle [139,140,141][111][112][113]. VSMCs expressing macrophage markers are also present in AAAs. [27,118][29][114].4.2.2. Senescent VSMCs
Chronic inflammation of the vessel wall is bound to cause the death of VSMCs, and the depletion of contractile VSMCs is a hallmark of AAAs. The scRNA-seq analyses failed to identify any proliferating VSMCs in the AAA specimens, indicating the failure of the VSMCs to activate into a proliferative phenotype. Nevertheless, a group of VSMCs that specifically express high levels of GATA6 and undergo glycolytic reprogramming has been identified [118][114]. GATA6 plays a multifaceted role in regulating VSMC phenotype. GATA6 has been implicated in the differentiation of VSMCs, as studies have shown that specific knockout of GATA6 in VSMCs can activate the expression of synthetic markers [147,148][115][116].4.2.3. Stressed VSMCs
Stress events such as oxidative stress, mitochondrial dysfunction, and unfolded protein response have been shown to alter the VSMC phenotype toward pro-inflammation and proliferation [153,154,155,156][117][118][119][120]. A group of VSMCs with transcriptomic features similar to contractile VSMCs, except a characteristically high expression of stress-related genes (FOS, ATF3, JUN, and HSPB8), were identified in TAAs and annotated as stressed VSMCs [111][87]. VSMCs with contractile VSMC transcriptome and high FOS, Jun, Klf2, and ATF3 expression were also identified in elastase-induced AAAs [118][114]. Due to the relatively high expression of genes associated with proliferation, they were annotated as proliferative VSMCs by the other. However, the actual proliferative capacity of this group of cells is uncertain because they express a high level of mitogen-activated protein kinase phosphatase-1 (Dusp1), which inhibits proliferation by suppressing MAPK activity.5. Role of VSMC Phenotypic Diversity in AAs
In clinical samples from patients with AAs, changes in VSMC phenotype affect hemostasis of the arterial wall, and VSMC plasticity has been correlated with disease severity. Such studies, however, represent only the final stage of the disease. Thus, whether VSMC heterogeneity and plasticity play a causative or compensatory role in AAs remains controversial. Animal models are tools for studying VSMC pathogenesis and phenotypic disruption of VSMCs can be inferred to cause hereditary aortic aneurysms from genetically engineered models [62,66,157][54][121][122]. VSMCs of different lineages respond differently to the same genetic mutation, and SHF-derived VSMCs are more phenotypically vulnerable than CNC-derived VSMCs. In contrast to hereditary AA, sporadic AA is a multifactorial disease that poses difficulties in the construction of animal models. At present, no animal model perfectly mimics human non-genetic AAs [164,165,166][123][124][125]. Results from non-hereditary models of AA support to some extent, the compensatory role of VSMC phenotypic transformation. To induce AA in WT mice, a minimum of two risk factors as well as sufficient challenge time are required. Challenging the aorta with a single risk factor such as Ang II infusion, hypertension, or hypercholesterolemia alone can elicit phenotypic changes of VSMCs but is not insufficient to induce AAs [105,106,107,108,109,110][81][82][83][84][85][86]. Extreme agents such as calcium chloride and elastase, which directly destroy the aortic wall, do induce AAs [167,168,169][126][127][128]. Despite the potential protective effects of the phenotypic alteration of VSMCs, the development and progression of sporadic AAs are indicative of maladaptation. The scholars propose that the sustained presence of noxious stimuli causes a prolonged phenotypic transformation of VSMCs, which causes pathological remodeling of the aortic wall and eventually leads to non-hereditary AAs. Proteoglycan and collagen accumulation prevent aortic dissection and rupture, however, overaccumulation of proteoglycans and collagen results in stiffening of the aortic wall which disrupts the phenotypic change of VSMCs [171,172][129][130]. Elevated ECM stiffness has been reported to polarize contractile VSMCs toward proinflammatory phenotypes through mechanotransduction [173,174,175,176][131][132][133][134].References
- Shen, Y.H.; LeMaire, S.A.; Webb, N.R.; Cassis, L.A.; Daugherty, A.; Lu, H.S. Aortic Aneurysms and Dissections Series. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e37–e46.
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606.
- Arnaud, P.; Hanna, N.; Benarroch, L.; Aubart, M.; Bal, L.; Bouvagnet, P.; Busa, T.; Dulac, Y.; Dupuis-Girod, S.; Edouard, T.; et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet Med. 2019, 21, 2015–2024.
- Bradley, D.; Badger, S.; McFarland, M.; Hughes, A. Abdominal Aortic Aneurysm Genetic Associations: Mostly False? A Systematic Review and Meta-analysis. Eur. J. Vasc. Endovasc. Surg. 2015, 51, 64–75.
- Dong, C.X.; Malecki, C.; Robertson, E.; Hambly, B.; Jeremy, R. Molecular Mechanisms in Genetic Aortopathy–Signaling Pathways and Potential Interventions. Int. J. Mol. Sci. 2023, 24, 1795.
- Davis, E.C. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab. Investig. 1993, 68, 89–99.
- Chamley-Campbell, J.H.; Campbell, G.R. What controls smooth muscle phenotype? Atherosclerosis 1981, 40, 347–357.
- Pyun, J.-H.; Ahn, B.-Y.; Vuong, T.A.; Kim, S.W.; Jo, Y.; Jeon, J.; Baek, S.H.; Kim, J.; Park, S.; Bae, G.-U.; et al. Inducible Prmt1 ablation in adult vascular smooth muscle leads to contractile dysfunction and aortic dissection. Exp. Mol. Med. 2021, 53, 1569–1579.
- Chiarini, A.; Onorati, F.; Marconi, M.; Pasquali, A.; Patuzzo, C.; Malashicheva, A.; Irtyega, O.; Faggian, G.; Pignatti, P.F.; Trabetti, E.; et al. Studies on sporadic non-syndromic thoracic aortic aneurysms: II. Alterations of extra-cellular matrix components and focal adhesion proteins. Eur. J. Prev. Cardiol. 2018, 25, 51–58.
- Chiarini, A.; Onorati, F.; Marconi, M.; Pasquali, A.; Patuzzo, C.; Malashicheva, A.; Irtyega, O.; Faggian, G.; Pignatti, P.F.; Trabetti, E.; et al. Studies on sporadic non-syndromic thoracic aortic aneurysms: 1. Deregulation of Jagged/Notch 1 homeostasis and selection of synthetic/secretor phenotype smooth muscle cells. Eur. J. Prev. Cardiol. 2018, 25, 42–50.
- Bochaton-Piallat, M.-L.; Bäck, M. Novel concepts for the role of smooth muscle cells in vascular disease: Towards a new smooth muscle cell classification. Cardiovasc. Res. 2018, 114, 477–480.
- Stenmark, K.R.; Frid, M.G.; Graham, B.B.; Tuder, R.M. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc. Res. 2018, 114, 551–564.
- Frid, M.G.; Dempsey, E.C.; Durmowicz, A.G.; Stenmark, K.R. Smooth Muscle Cell Heterogeneity in Pulmonary and Systemic Vessels. Importance in vascular disease. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1203–1209.
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.-L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550.
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164.
- Campbell, J.H.; Campbell, G.R. Smooth Muscle Phenotypic Modulation—A Personal Experience. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1784–1789.
- Majesky, M.W. Developmental Basis of Vascular Smooth Muscle Diversity. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1248–1258.
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90.
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616.
- Wasteson, P.; Johansson, B.R.; Jukkola, T.; Breuer, S.; Akyürek, L.M.; Partanen, J.; Lindahl, P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development 2008, 135, 1823–1832.
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth Muscle Cells Derived From Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1722–1726.
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.; Cochain, C.; Vafadarnejad, E.; Saliba, A.-E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688.
- Kalluri, A.S.; Vellarikkal, S.K.; Edelman, E.R.; Nguyen, L.; Subramanian, A.; Ellinor, P.T.; Regev, A.; Kathiresan, S.; Gupta, R.M. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 2019, 140, 147–163.
- Sugiura, T.; Freis, E.D. Pressure Pulse in Small Arteries. Circ. Res. 1962, 11, 838–842.
- Leuprecht, A.; Perktold, K.; Kozerke, S.; Boesiger, P. Combined CFD and MRI study of blood flow in a human ascending aorta model. Biorheology 2002, 39, 425–429.
- Vincent, P.E.; Plata, A.M.; Hunt, A.A.E.; Weinberg, P.D.; Sherwin, S.J. Blood flow in the rabbit aortic arch and descending thoracic aorta. J. R. Soc. Interface 2011, 8, 1708–1719.
- Stein, P.D.; Sabbah, H.N. Turbulent blood flow in the ascending aorta of humans with normal and diseased aortic valves. Circ. Res. 1976, 39, 58–65.
- Chandran, K.B. Flow Dynamics in the Human Aorta. J. Biomech. Eng. 1993, 115, 611–616.
- Yu, L.; Zhang, J.; Gao, A.; Zhang, M.; Wang, Z.; Yu, F.; Guo, X.; Su, G.; Zhang, Y.; Zhang, C. An intersegmental single-cell profile reveals aortic heterogeneity and identifies a novel Malat1+ vascular smooth muscle subtype involved in abdominal aortic aneurysm formation. Signal Transduct. Target. Ther. 2022, 7, 125.
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.B.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin–dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173.
- Gadson, P.F., Jr.; Dalton, M.L.; Patterson, E.; Svoboda, D.D.; Hutchinson, L.; Schram, D.; Rosenquist, T.H. Differential Response of Mesoderm- and Neural Crest-Derived Smooth Muscle to TGF-β1: Regulation of c-myb and α1 (I) Procollagen Genes. Exp. Cell Res. 1997, 230, 169–180.
- Topouzis, S.; Majesky, M.W. Smooth Muscle Lineage Diversity in the Chick Embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta. Dev. Biol. 1996, 178, 430–445.
- Thieszen, S.L.; Dalton, M.; Gadson, P.F.; Patterson, E.; Rosenquist, T.H. Embryonic Lineage of Vascular Smooth Muscle Cells Determines Responses to Collagen Matrices and Integrin Receptor Expression. Exp. Cell Res. 1996, 227, 135–145.
- Oyama, N.; Gona, P.; Salton, C.J.; Chuang, M.L.; Jhaveri, R.R.; Blease, S.J.; Manning, A.R.; Lahiri, M.; Botnar, R.M.; Levy, D.; et al. Differential Impact of Age, Sex, and Hypertension on Aortic Atherosclerosis: The Framingham Heart Study. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 155–159.
- Neisius, U.; Gona, P.N.; Oyama-Manabe, N.; Chuang, M.L.; O’donnell, C.J.; Manning, W.J.; Tsao, C.W. Relation of MRI Aortic Wall Area and Plaque to Incident Cardiovascular Events: The Framingham Heart Study. Radiology 2022, 304, 542–550.
- Haimovici, H.; Maier, N. Fate of aortic homografts in canine atherosclerosis. 3. study of fresh abdominal and thoracic aortic implants into thoracic aorta: Role of tissue susceptibility in atherogenesis. Arch. Surg. 1964, 89, 961–969.
- Leroux-Berger, M.; Queguiner, I.; Maciel, T.T.; Ho, A.; Relaix, F.; Kempf, H. Pathologic calcification of adult vascular smooth muscle cells differs on their crest or mesodermal embryonic origin. J. Bone Miner. Res. 2011, 26, 1543–1553.
- Muyor, K.; Laget, J.; Cortijo, I.; Duranton, F.; Jover, B.; Argilés, À.; Gayrard, N. Vascular calcification in different arterial beds in ex vivo ring culture and in vivo rat model. Sci. Rep. 2022, 12, 11861.
- Li, Y.; Fang, M.; Yang, J.; Yu, C.; Kuang, J.; Sun, T.; Fan, R. Analysis of the contribution of 129 candidate genes to thoracic aortic aneurysm or dissection of a mixed cohort of sporadic and familial cases in South China. Am. J. Transl. Res. 2021, 13, 4281–4295.
- Isselbacher, E.M.; Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528.
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615.
- Sakai, L.Y.; Keene, D.R. Fibrillin protein pleiotropy: Acromelic dysplasias. Matrix Biol. 2019, 80, 6–13.
- Prakash, S.K.; LeMaire, S.A.; Guo, D.-C.; Russell, L.; Regalado, E.S.; Golabbakhsh, H.; Johnson, R.J.; Safi, H.J.; Estrera, A.L.; Coselli, J.S.; et al. Rare Copy Number Variants Disrupt Genes Regulating Vascular Smooth Muscle Cell Adhesion and Contractility in Sporadic Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2010, 87, 743–756.
- Guo, J.; Cai, L.; Jia, L.; Li, X.; Xi, X.; Zheng, S.; Liu, X.; Piao, C.; Liu, T.; Sun, Z.; et al. Wide mutation spectrum and frequent variant Ala27Thr of FBN1 identified in a large cohort of Chinese patients with sporadic TAAD. Sci. Rep. 2015, 5, 13115.
- Li, Y.; Gao, S.; Han, Y.; Song, L.; Kong, Y.; Jiao, Y.; Huang, S.; Du, J.; Li, Y. Variants of Focal Adhesion Scaffold Genes Cause Thoracic Aortic Aneurysm. Circ. Res. 2021, 128, 8–23.
- Liu, C.-L.; Liu, X.; Zhang, Y.; Liu, J.; Yang, C.; Luo, S.; Liu, T.; Wang, Y.; Lindholt, J.S.; Diederichsen, A.; et al. Eosinophils Protect Mice From Angiotensin-II Perfusion–Induced Abdominal Aortic Aneurysm. Circ. Res. 2021, 128, 188–202.
- Benjamins, J.W.; Yeung, M.W.; van de Vegte, Y.J.; Said, M.A.; van der Linden, T.; Ties, D.; Juarez-Orozco, L.E.; Verweij, N.; van der Harst, P. Genomic insights in ascending aortic size and distensibility. Ebiomedicine 2022, 75, 103783.
- Milewicz, D.M.; Braverman, A.C.; De Backer, J.; Morris, S.A.; Boileau, C.; Maumenee, I.H.; Jondeau, G.; Evangelista, A.; Pyeritz, R.E. Marfan syndrome. Nat. Rev. Dis. Primers 2021, 7, 64.
- Van Laer, L.; Dietz, H.; Loeys, B. Loeys-Dietz syndrome. Adv. Exp. Med. Biol. 2014, 802, 95–105.
- Gouda, P.; Kay, R.; Habib, M.; Aziz, A.; Aziza, E.; Welsh, R. Clinical features and complications of Loeys-Dietz syndrome: A systematic review. Int. J. Cardiol. 2022, 362, 158–167.
- Schoenhoff, F.S.; Alejo, D.E.; Black, J.H.; Crawford, T.C.; Dietz, H.C.; Grimm, J.C.; Magruder, J.T.; Patel, N.D.; Vricella, L.A.; Young, A.; et al. Management of the aortic arch in patients with Loeys–Dietz syndrome. J. Thorac. Cardiovasc. Surg. 2020, 160, 1166–1175.
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594.
- MacFarlane, E.G.; Parker, S.J.; Shin, J.Y.; Ziegler, S.G.; Creamer, T.J.; Bagirzadeh, R.; Bedja, D.; Chen, Y.; Calderon, J.F.; Weissler, K.; et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J. Clin. Investig. 2019, 129, 659–675.
- Zhou, D.; Feng, H.; Yang, Y.; Huang, T.; Qiu, P.; Zhang, C.; Olsen, T.R.; Zhang, J.; Chen, Y.E.; Mizrak, D.; et al. hiPSC Modeling of Lineage-Specific Smooth Muscle Cell Defects Caused by TGFBR1 A230T Variant, and Its Therapeutic Implications for Loeys-Dietz Syndrome. Circulation 2021, 144, 1145–1159.
- Jaffe, M.; Sesti, C.; Washington, I.M.; Du, L.; Dronadula, N.; Chin, M.T.; Stolz, D.B.; Davis, E.C.; Dichek, D.A. Transforming Growth Factor-β Signaling in Myogenic Cells Regulates Vascular Morphogenesis, Differentiation, and Matrix Synthesis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e1–e11.
- Angelov, S.N.; Hu, J.H.; Wei, H.; Airhart, N.; Shi, M.; Dichek, D.A. TGF-β (Transforming Growth Factor-β) Signaling Protects the Thoracic and Abdominal Aorta From Angiotensin II-Induced Pathology by Distinct Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2102–2113.
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Investig. 2014, 124, 755–767.
- Gittenbergerdegroot, A.; Azhar, M.; Molin, D. Transforming Growth Factor β–SMAD2 Signaling and Aortic Arch Development. Trends Cardiovasc. Med. 2006, 16, 1–6.
- Molin, D.G.; Poelmann, R.E.; DeRuiter, M.C.; Azhar, M.; Doetschman, T.; Groot, A.C.G.-D. Transforming Growth Factor β–SMAD2 Signaling Regulates Aortic Arch Innervation and Development. Circ. Res. 2004, 95, 1109–1117.
- Langlois, D.; Hneino, M.; Bouazza, L.; Parlakian, A.; Sasaki, T.; Bricca, G.; Li, J.Y. Conditional inactivation of TGF-β type II receptor in smooth muscle cells and epicardium causes lethal aortic and cardiac defects. Transgenic Res. 2010, 19, 1069–1082.
- Wurdak, H.; Ittner, L.M.; Lang, K.S.; Leveen, P.; Suter, U.; Fischer, J.A.; Karlsson, S.; Born, W.; Sommer, L. Inactivation of TGFβ signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005, 19, 530–535.
- Choudhary, B.; Ito, Y.; Makita, T.; Sasaki, T.; Chai, Y.; Sucov, H.M. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFβ receptor (Tgfbr2) mutant mice. Dev. Biol. 2006, 289, 420–429.
- Zhu, J.; Angelov, S.; Yildirim, I.A.; Wei, H.; Hu, J.H.; Majesky, M.W.; Brozovich, F.V.; Kim, F.; Dichek, D.A. Loss of Transforming Growth Factor Beta Signaling in Aortic Smooth Muscle Cells Causes Endothelial Dysfunction and Aortic Hypercontractility. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1956–1971.
- Xie, W.-B.; Li, Z.; Shi, N.; Guo, X.; Tang, J.; Ju, W.; Han, J.; Liu, T.; Bottinger, E.P.; Chai, Y.; et al. Smad2 and Myocardin-Related Transcription Factor B Cooperatively Regulate Vascular Smooth Muscle Differentiation From Neural Crest Cells. Circ. Res. 2013, 113, e76–e86.
- Huang, W.-Y.; Xie, W.; Guo, X.; Li, F.; Jose, P.A.; Chen, S.-Y.; Jiang, H.; Chen, Y.; Yu, T.; Zhao, X.; et al. Smad2 and PEA3 cooperatively regulate transcription of response gene to complement 32 in TGF-β-induced smooth muscle cell differentiation of neural crest cells. Am. J. Physiol. Cell Physiol. 2011, 301, C499–C506.
- Inamoto, S.; Kwartler, C.S.; Lafont, A.L.; Liang, Y.Y.; Fadulu, V.T.; Duraisamy, S.; Willing, M.; Estrera, A.; Safi, H.; Hannibal, M.C.; et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc. Res. 2010, 88, 520–529.
- Ferruzzi, J.; Murtada, S.-I.; Li, G.; Jiao, Y.; Uman, S.; Ting, M.Y.L.; Tellides, G.; Humphrey, J.D. Pharmacologically Improved Contractility Protects against Aortic Dissection in Mice with Disrupted Transforming Growth Factor-β Signaling Despite Compromised Extracellular Matrix Properties. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 919–927.
- Robertson, I.; Jensen, S.; Handford, P. TB domain proteins: Evolutionary insights into the multifaceted roles of fibrillins and LTBPs. Biochem. J. 2011, 433, 263–276.
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic Alteration of Vascular Smooth Muscle Cells Precedes Elastolysis in a Mouse Model of Marfan Syndrome. Circ. Res. 2001, 88, 37–43.
- Iosef, C.; Pedroza, A.J.; Cui, J.Z.; Dalal, A.R.; Arakawa, M.; Tashima, Y.; Koyano, T.K.; Burdon, G.; Churovich, S.M.P.; Orrick, J.O.; et al. Quantitative proteomics reveal lineage-specific protein profiles in iPSC-derived Marfan syndrome smooth muscle cells. Sci. Rep. 2020, 10, 20392.
- Elango, J.; Hou, C.; Bao, B.; Wang, S.; de Val, J.E.M.S.; Wenhui, W. The Molecular Interaction of Collagen with Cell Receptors for Biological Function. Polymers 2022, 14, 876.
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43.
- Yamashiro, Y.; Thang, B.Q.; Shin, S.J.; Lino, C.A.; Nakamura, T.; Kim, J.; Sugiyama, K.; Tokunaga, C.; Sakamoto, H.; Osaka, M.; et al. Role of Thrombospondin-1 in Mechanotransduction and Development of Thoracic Aortic Aneurysm in Mouse and Humans. Circ. Res. 2018, 123, 660–672.
- Liu, Z.; Morgan, S.; Ren, J.; Wang, Q.; Annis, D.S.; Mosher, D.F.; Zhang, J.; Sorenson, C.M.; Sheibani, N.; Liu, B.; et al. Thrombospondin-1 (TSP1) Contributes to the Development of Vascular Inflammation by Regulating Monocytic Cell Motility in Mouse Models of Abdominal Aortic Aneurysm. Circ. Res. 2015, 117, 129–141.
- Li, H.; Xu, H.; Wen, H.; Wang, H.; Zhao, R.; Sun, Y.; Bai, C.; Ping, J.; Song, L.; Luo, M.; et al. Lysyl hydroxylase 1 (LH1) deficiency promotes angiotensin II (Ang II)-induced dissecting abdominal aortic aneurysm. Theranostics 2021, 11, 9587–9604.
- Yang, H.; Zhou, T.; Sorenson, C.M.; Sheibani, N.; Liu, B. Myeloid-Derived TSP1 (Thrombospondin-1) Contributes to Abdominal Aortic Aneurysm Through Suppressing Tissue Inhibitor of Metalloproteinases-1. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e350–e366.
- Pedroza, A.J.; Tashima, Y.; Shad, R.; Cheng, P.; Wirka, R.; Churovich, S.; Nakamura, K.; Yokoyama, N.; Cui, J.Z.; Iosef, C.; et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2195–2211.
- Pedroza, A.J.; Dalal, A.R.; Shad, R.; Yokoyama, N.; Nakamura, K.; Cheng, P.; Wirka, R.C.; Mitchel, O.; Baiocchi, M.; Hiesinger, W.; et al. Embryologic Origin Influences Smooth Muscle Cell Phenotypic Modulation Signatures in Murine Marfan Syndrome Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1154–1168.
- Pyeritz, R.E. Heritable thoracic aortic disorders. Curr. Opin. Cardiol. 2014, 29, 97–102.
- Robertson, E.N.; Hambly, B.D.; Jeremy, R.W. Thoracic aortic dissection and heritability: Forensic implications. Forensic Sci. Med. Pathol. 2016, 12, 366–368.
- Lu, H.; Daugherty, A. Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e59–e65.
- Kan, H.; Zhang, K.; Mao, A.; Geng, L.; Gao, M.; Feng, L.; You, Q.; Ma, X. Single-cell transcriptome analysis reveals cellular heterogeneity in the ascending aortas of normal and high-fat diet-fed mice. Exp. Mol. Med. 2021, 53, 1379–1389.
- Zhang, K.; Kan, H.; Mao, A.; Geng, L.; Ma, X. Single-cell analysis of salt-induced hypertensive mouse aortae reveals cellular heterogeneity and state changes. Exp. Mol. Med. 2021, 53, 1866–1876.
- Salmon, M.; Johnston, W.F.; Woo, A.; Pope, N.H.; Su, G.; Upchurch, G.R., Jr.; Owens, G.K.; Ailawadi, G. KLF4 Regulates Abdominal Aortic Aneurysm Morphology and Deletion Attenuates Aneurysm Formation. Circulation 2013, 128, S163–S174.
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637.
- Cheng, J.; Gu, W.; Lan, T.; Deng, J.; Ni, Z.; Zhang, Z.; Hu, Y.; Sun, X.; Yang, Y.; Xu, Q. Single-cell RNA sequencing reveals cell type- and artery type-specific vascular remodelling in male spontaneously hypertensive rats. Cardiovasc. Res. 2021, 117, 1202–1216.
- Li, Y.; Ren, P.; Dawson, A.; Vasquez, H.G.; Ageedi, W.; Zhang, C.; Luo, W.; Chen, R.; Li, Y.; Kim, S.; et al. Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation 2020, 142, 1374–1388.
- Cao, G.; Lu, Z.; Gu, R.; Xuan, X.; Zhang, R.; Hu, J.; Dong, H. Deciphering the Intercellular Communication Between Immune Cells and Altered Vascular Smooth Muscle Cell Phenotypes in Aortic Aneurysm From Single-Cell Transcriptome Data. Front. Cardiovasc. Med. 2022, 9, 936287.
- Song, W.; Qin, L.; Chen, Y.; Chen, J.; Wei, L. Single-cell transcriptome analysis identifies Versican(+) myofibroblast as a hallmark for thoracic aortic aneurysm marked by activation of PI3K-AKT signaling pathway. Biochem. Biophys. Res. Commun. 2022, 643, 175–185.
- Chou, E.L.; Chaffin, M.; Simonson, B.; Pirruccello, J.P.; Akkad, A.-D.; Nekoui, M.; Cardenas, C.L.L.; Bedi, K.C.; Nash, C.; Juric, D.; et al. Aortic Cellular Diversity and Quantitative Genome-Wide Association Study Trait Prioritization Through Single-Nuclear RNA Sequencing of the Aneurysmal Human Aorta. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1355–1374.
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican—A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512.
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289.
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075.
- Ma, Q.; Yang, Q.; Xu, J.; Zhang, X.; Kim, D.; Liu, Z.; Da, Q.; Mao, X.; Zhou, Y.; Cai, Y.; et al. ATIC-Associated De Novo Purine Synthesis Is Critically Involved in Proliferative Arterial Disease. Circulation 2022, 146, 1444–1460.
- Majesky, M.W.; Horita, H.; Ostriker, A.; Lu, S.; Regan, J.N.; Bagchi, A.; Dong, X.R.; Poczobutt, J.; Nemenoff, R.A.; Weiser-Evans, M.C.; et al. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4. Circ. Res. 2017, 120, 296–311.
- Shikatani, E.A.; Chandy, M.; Besla, R.; Li, C.C.; Momen, A.; El-Mounayri, O.; Robbins, C.S.; Husain, M. c-Myb Regulates Proliferation and Differentiation of Adventitial Sca1 + Vascular Smooth Muscle Cell Progenitors by Transactivation of Myocardin. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1367–1376.
- Worssam, M.D.; Lambert, J.; Oc, S.; Taylor, J.C.K.; Taylor, A.L.; Dobnikar, L.; Chappell, J.; Harman, J.L.; Figg, N.L.; Finigan, A.; et al. Cellular mechanisms of oligoclonal vascular smooth muscle cell expansion in cardiovascular disease. Cardiovasc. Res. 2022, 119, 1279–1294.
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jørgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323.
- Chen, P.-Y.; Qin, L.; Li, G.; Malagon-Lopez, J.; Wang, Z.; Bergaya, S.; Gujja, S.; Caulk, A.W.; Murtada, S.-I.; Zhang, X.; et al. Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell 2020, 26, 542–557.e511.
- Clément, M.; Chappell, J.; Raffort, J.; Lareyre, F.; Vandestienne, M.; Taylor, A.L.; Finigan, A.; Harrison, J.; Bennett, M.R.; Bruneval, P.; et al. Vascular Smooth Muscle Cell Plasticity and Autophagy in Dissecting Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1149–1159.
- Zhang, C.; Li, Y.; Chakraborty, A.; Li, Y.; Rebello, K.R.; Ren, P.; Luo, W.; Zhang, L.; Lu, H.S.; Cassis, L.A.; et al. Aortic Stress Activates an Adaptive Program in Thoracic Aortic Smooth Muscle Cells That Maintains Aortic Strength and Protects against Aneurysm and Dissection in Mice. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 234–252.
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis. Circ. Res. 2014, 115, 662–667.
- Harman, J.L.; Jørgensen, H.F. The role of smooth muscle cells in plaque stability: Therapeutic targeting potential. Br. J. Pharmacol. 2019, 176, 3741–3753.
- Li, Y.; Zhu, H.; Zhang, Q.; Han, X.; Zhang, Z.; Shen, L.; Wang, L.; Lui, K.O.; He, B.; Zhou, B. Smooth muscle-derived macrophage-like cells contribute to multiple cell lineages in the atherosclerotic plaque. Cell Discov. 2021, 7, 111.
- Leone, O.; Corsini, A.; Pacini, D.; Corti, B.; Lorenzini, M.; Laus, V.; Foà, A.; Reggiani, M.L.B.; Di Marco, L.; Rapezzi, C. The complex interplay among atherosclerosis, inflammation, and degeneration in ascending thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2019, 160, 1434–1443.e6.
- Nesi, G.; Anichini, C.; Tozzini, S.; Boddi, V.; Calamai, G.; Gori, F. Pathology of the thoracic aorta: A morphologic review of 338 surgical specimens over a 7-year period. Cardiovasc. Pathol. 2009, 18, 134–139.
- He, Y.-B.; Jin, H.-Z.; Zhao, J.-L.; Wang, C.; Ma, W.-R.; Xing, J.; Zhang, X.-B.; Zhang, Y.-Y.; Dai, H.-D.; Zhao, N.-S.; et al. Single-cell transcriptomic analysis reveals differential cell subpopulations and distinct phenotype transition in normal and dissected ascending aorta. Mol. Med. 2022, 28, 158.
- Negedu, M.N.; Duckworth, C.A.; Yu, L.-G. Galectin-2 in Health and Diseases. Int. J. Mol. Sci. 2022, 24, 341.
- Minas, T.Z.; Candia, J.; Dorsey, T.H.; Baker, F.; Tang, W.; Kiely, M.; Smith, C.J.; Zhang, A.L.; Jordan, S.V.; Obadi, O.M.; et al. Serum proteomics links suppression of tumor immunity to ancestry and lethal prostate cancer. Nat. Commun. 2022, 13, 1759.
- Huang, Y.; Wang, H.-C.; Zhao, J.; Wu, M.-H.; Shih, T.-C. Immunosuppressive Roles of Galectin-1 in the Tumor Microenvironment. Biomolecules 2021, 11, 1398.
- López-Otín, C.; Kroemer, G. Hallmarks of Health. Cell 2021, 184, 33–63.
- Luo, W.; Wang, Y.; Zhang, L.; Ren, P.; Zhang, C.; Li, Y.; Azares, A.R.; Zhang, M.; Guo, J.; Ghaghada, K.B.; et al. Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture. Circulation 2020, 141, 42–66.
- Saito, T.; Hasegawa, Y.; Ishigaki, Y.; Yamada, T.; Gao, J.; Imai, J.; Uno, K.; Kaneko, K.; Ogihara, T.; Shimosawa, T.; et al. Importance of endothelial NF-κB signalling in vascular remodelling and aortic aneurysm formation. Cardiovasc. Res. 2013, 97, 106–114.
- Zhao, G.; Lu, H.; Chang, Z.; Zhao, Y.; Zhu, T.; Chang, L.; Guo, Y.; Garcia-Barrio, M.T.; Chen, Y.E.; Zhang, J. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc. Res. 2020, 117, 1402–1416.
- Kurz, J.; Weiss, A.-C.; Lüdtke, T.H.-W.; Deuper, L.; Trowe, M.-O.; Thiesler, H.; Hildebrandt, H.; Heineke, J.; Duncan, S.A.; Kispert, A. GATA6 is a crucial factor for Myocd expression in the visceral smooth muscle cell differentiation program of the murine ureter. Development 2022, 149, dev200522.
- Zhuang, T.; Liu, J.; Chen, X.; Pi, J.; Kuang, Y.; Wang, Y.; Tomlinson, B.; Chan, P.; Zhang, Q.; Li, Y.; et al. Cell-Specific Effects of GATA (GATA Zinc Finger Transcription Factor Family)-6 in Vascular Smooth Muscle and Endothelial Cells on Vascular Injury Neointimal Formation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 888–901.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell. Mol. Life Sci. 2016, 73, 2405–2410.
- Chattopadhyay, A.; Kwartler, C.S.; Kaw, K.; Li, Y.; Kaw, A.; Chen, J.; LeMaire, S.A.; Shen, Y.H.; Milewicz, D.M. Cholesterol-Induced Phenotypic Modulation of Smooth Muscle Cells to Macrophage/Fibroblast–like Cells Is Driven by an Unfolded Protein Response. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 302–316.
- Hamczyk, M.R.; Villa-Bellosta, R.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Andrés-Manzano, M.J.; Misteli, T.; López-Otín, C.; Andrés, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11, e9736.
- Oller, J.; Gabandé-Rodríguez, E.; Ruiz-Rodríguez, M.J.; Desdín-Micó, G.; Aranda, J.F.; Rodrigues-Diez, R.; Ballesteros-Martínez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuña, P.; et al. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 2021, 143, 2091–2109.
- Milewicz, D.M.; Trybus, K.M.; Guo, D.-C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34.
- Asano, K.; Cantalupo, A.; Sedes, L.; Ramirez, F. Pathophysiology and Therapeutics of Thoracic Aortic Aneurysm in Marfan Syndrome. Biomolecules 2022, 12, 128.
- Krishna, S.M.; Morton, S.K.; Li, J.; Golledge, J. Risk Factors and Mouse Models of Abdominal Aortic Aneurysm Rupture. Int. J. Mol. Sci. 2020, 21, 7250.
- Lu, G.; Su, G.; Davis, J.P.; Schaheen, B.; Downs, E.; Roy, R.J.; Ailawadi, G.; Upchurch, G.R. A novel chronic advanced stage abdominal aortic aneurysm murine model. J. Vasc. Surg. 2017, 66, 232–242.e4.
- Golledge, J.; Krishna, S.M.; Wang, Y. Mouse models for abdominal aortic aneurysm. Br. J. Pharmacol. 2020, 179, 792–810.
- Sulé-Suso, J.; Forster, A.; Zholobenko, V.; Stone, N.; El Haj, A. Effects of CaCl2 and MgCl2 on Fourier Transform Infrared Spectra of Lung Cancer Cells. Appl. Spectrosc. 2004, 58, 61–67.
- Sundararaman, S.S.; van der Vorst, E.P.C. Calcium-Sensing Receptor (CaSR), Its Impact on Inflammation and the Consequences on Cardiovascular Health. Int. J. Mol. Sci. 2021, 22, 2478.
- Wagenseil, J.E.; Mecham, R.P.; Tellides, G.; Staiculescu, M.C.; Cocciolone, A.J.; Procknow, J.D.; Kim, J.; Hawes, J.Z.; Johnson, E.O.; Murshed, M.; et al. Vascular Extracellular Matrix and Arterial Mechanics. Physiol. Rev. 2009, 89, 957–989.
- Wong, L.; Kumar, A.; Gabela-Zuniga, B.; Chua, J.; Singh, G.; Happe, C.L.; Engler, A.J.; Fan, Y.; McCloskey, K.E. Substrate stiffness directs diverging vascular fates. Acta Biomater. 2019, 96, 321–329.
- Schnellmann, R.; Ntekoumes, D.; Choudhury, M.I.; Sun, S.; Wei, Z.; Gerecht, S. Stiffening Matrix Induces Age-Mediated Microvascular Phenotype Through Increased Cell Contractility and Destabilization of Adherens Junctions. Adv. Sci. 2022, 9, e2201483.
- Talwar, S.; Kant, A.; Xu, T.; Shenoy, V.B.; Assoian, R.K. Mechanosensitive smooth muscle cell phenotypic plasticity emerging from a null state and the balance between Rac and Rho. Cell Rep. 2021, 35, 109019.
- Wang, J.; Xie, S.-A.; Li, N.; Zhang, T.; Yao, W.; Zhao, H.; Pang, W.; Han, L.; Liu, J.; Zhou, J. Matrix stiffness exacerbates the proinflammatory responses of vascular smooth muscle cell through the DDR1-DNMT1 mechanotransduction axis. Bioact. Mater. 2022, 17, 406–424.
- Qian, W.; Hadi, T.; Silvestro, M.; Ma, X.; Rivera, C.F.; Bajpai, A.; Li, R.; Zhang, Z.; Qu, H.; Tellaoui, R.S.; et al. Microskeletal stiffness promotes aortic aneurysm by sustaining pathological vascular smooth muscle cell mechanosensation via Piezo1. Nat. Commun. 2022, 13, 512.
- Pasta, S.; Agnese, V.; Gallo, A.; Cosentino, F.; Di Giuseppe, M.; Gentile, G.; Raffa, G.M.; Maalouf, J.F.; Michelena, H.I.; Bellavia, D.; et al. Shear Stress and Aortic Strain Associations with Biomarkers of Ascending Thoracic Aortic Aneurysm. Ann. Thorac. Surg. 2020, 110, 1595–1604.