As the major cellular component of aortic walls, vascular smooth muscle cells (VSMCs) ought to be dynamic to adapt to the fluctuating microenvironment and to maintain the intact structure and functionality of the aortic wall
[6]. Unlike terminally differentiated cells, VSMCs can change their morphological and functional characteristics under certain conditions. VSMCs isolated from porcine aortas in sparse culture were observed to switch from a spindle to a polymorphic shape and exhibit logarithmic growth responses to mitogens
[7]. Years of research have led to the contractile-synthetic binary phenotype theory of VSMCs. It is believed that VSMCs have 2 distinct but interchangeable phenotypes. The contractile VSMCs, which contribute to the maintenance of vascular tone, express high levels of contraction-related genes, including
Acta2,
Myh11,
Tagln, and
Cnn1. In contrast, synthetic VSMCs are characterized by reduced expression of contraction-related genes expression and increased cell proliferation, migration, inflammation, and extracellular matrix (ECM) synthesis
[1][7][8][9][10][11][12][13][14][15][16][1,7,8,9,10,11,12,13,14,15,16]. According to the theory, healthy aortas contain only contractile VSMCs, and in pathological conditions, contractile VSMCs are transformed into synthetic VSMCs.
2. VSMC Heterogeneity in Normal Aorta
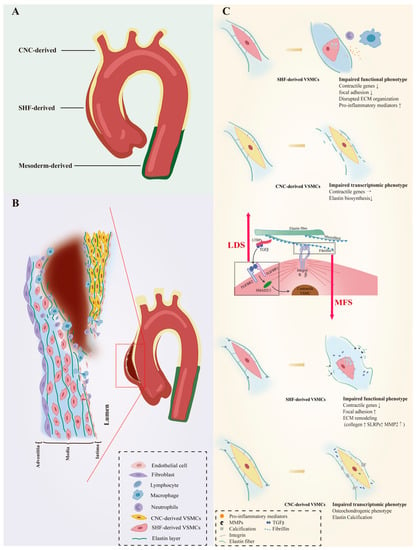
The unique embryonic context is a source of VSMC heterogeneity. As reviewed by Majesky, fate mapping technology in developing embryos has revealed a mosaic distribution of VSMCs from different precursor sources in the aorta, which includes VSMCs derived from the secondary heart fields (SHF), cardiac neural crest (CNC), somite, and splanchnic mesoderm
[17][19]. For instance, VSMCs of the aortic root are mainly of SHF origin, those of the ascending aorta and aortic arch originate mainly from the CNC, and VSMCs composing the descending aorta are derived from mesoderm
[18][19][20][20,21,22]. Even the same aortic segment can consist of VSMCs of different embryonic origins. SHF-derived VSMCs were previously thought to be restricted in the aortic root, forming a suture with CNC-derived VSMCs at the transition from the aortic root and the ascending aorta. However, lineage tracing shows that SHF-derived VSMCs extend distally to the innominate artery orifice. The ascending aorta actually contains both SHF-derived and CNC-derived VSMCs, with the SHF-derived VSMCs wrapping around the outside of the CNC-derived VSMCs in a sleeve-shaped form (
Figure 1A)
[18][21][20,23].
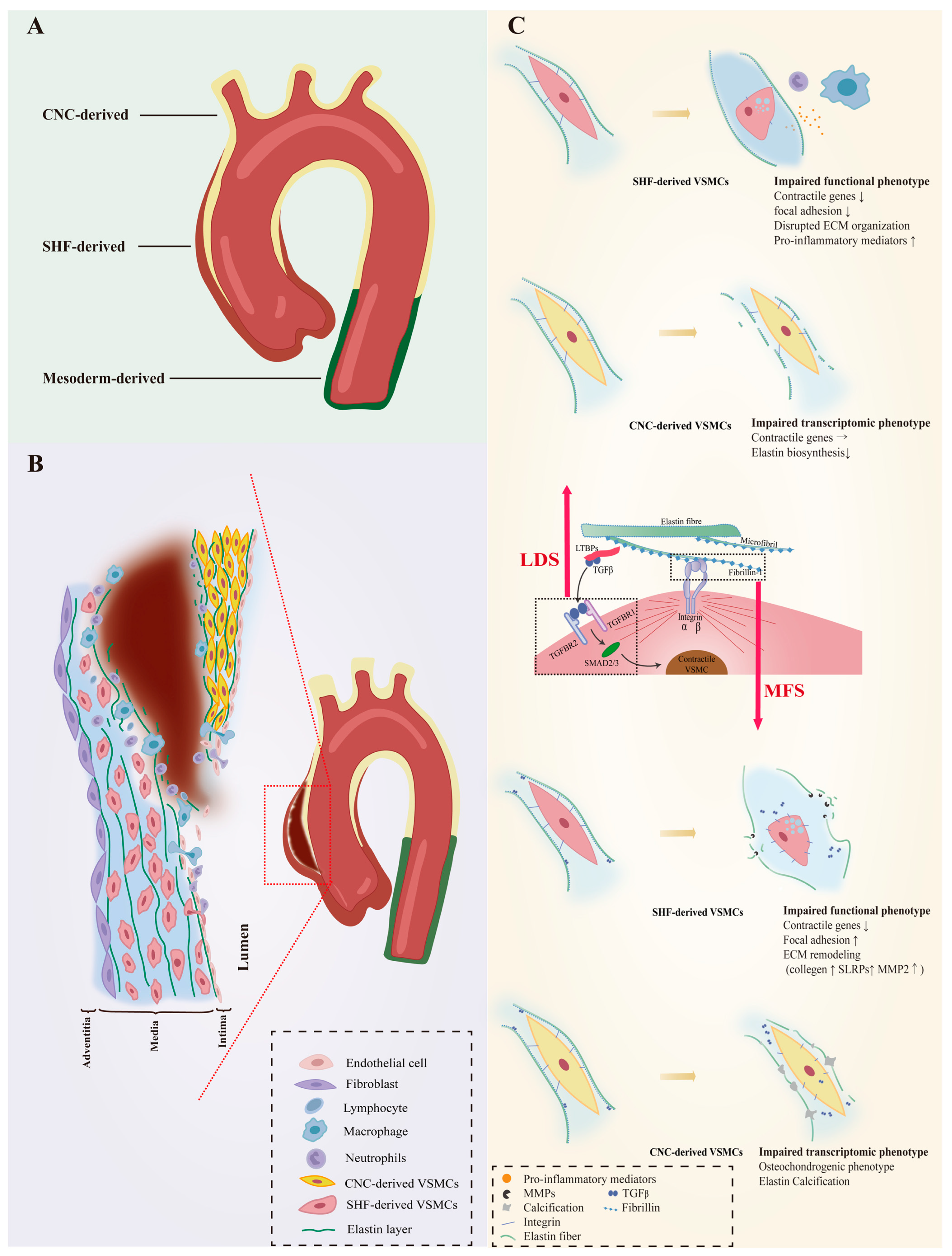
Figure 1. VSMC phenotypic diversity in hereditary AAs. The aortic wall consists of VSMCs from different embryonic origins that respond unevenly to genetic defects that directly or indirectly regulate VSMC contractile phenotype, which results in heterogeneous susceptibility to aortic diseases. (A) provides an overview of the origin of VSMCs in the aortic wall. The ascending aorta is composed of VSMCs derived from SHF and CNC. The former wraps outside of the latter in a “sleeve-shaped” form, However, mesoderm-derived VSMCs predominate in the abdominal aorta. (B) depicts a plausible hypothesis that the ascending aorta is susceptible to AAs and ADs when genetic defects are present. VSMCs with distinct embryonic backgrounds show different responses to signaling perturbations. Compared to CNC-derived VSMCs, SHF-derived VSMCs are more prone to lose their contractile phenotype and cause pathological ECM remodeling. This leads to a reduction in the strength of the aortic wall, making it susceptible to swelling. Moreover, the turbulent blood flow can easily cause tearing along the interface between the 2 VSMCs, leading to the occurrence of ADs in the presence of damaged endothelium. (C) shows the dysfunctional phenotypes of VSMCs from different origins with genetic defects. Fibrillin-1 is the primary protein found in the microfibril extensions of elastic lamellae, which are attached to focal adhesions on the surface of VSMCs. It is also associated with TGF-β binding proteins (LTBP), which are involved in the TGF-β signaling pathway. Genetic defects in fibrillin-1 (MFS) and defects in the TGF-β signaling pathway (LDS) affect the phenotype of both VSMCs, with SHF-derived VSMCs being more susceptible to loss of the contractile phenotype. The direction of arrows indicates the trend of gene expression levels: upward represent upregulation, horizontal indicate stable expression levels, and downward indicate downregulation. Vascular smooth muscle cells, VSMCs; Secondary heart field, SHF; Cardiac neural crest, CNC; Extracellular matrix, ECM; Aortic aneurysms, AAs; Aortic dissections, ADs; Loeys-Dietz syndrome, LDS; Marfan syndrome, MFS.
According to classical theory, lineage heterogeneous VSMCs have a common developmental fate that converts to a contractile phenotype, but the scRNA-seq results reveal richer VSMC diversity than previously thought. Despite being the predominant cell type of the aorta, VSMCs have received much less attention than immune cells and endothelial cells in studies to obtain single-cell ATLAS of the aortic wall. Therefore, most of the published results simply identify VSMCs based on canonical VSMC markers without doing sub-clustering
[22][23][24,25].
The heterogeneity of VSMCs may be a physiological adaptation. Hemodynamics vary in different segments of the aorta. Blood velocity in the ascending aorta and aortic arch is faster than in the abdominal aorta, causing greater vessel wall impact
[24][25][33,34]. Due to the anatomical curvature, the blood flow in the ascending aorta, especially in the aortic arch, changes drastically, resulting in vortex flow and increased demands on the strength and self-healing ability of the vascular wall
[26][27][28][35,36,37]. Vim-expressing VSMCs enriched for genes associated with shear stress, atherogenesis, WNT, and MAPK pathways populate the thoracic aorta and aortic arch, representing a group of flow-shock-adapted VSMCs that contribute to higher elastin/collagen content
[29][27]. Interestingly, the regional distribution of VSMCs from different embryologic backgrounds is consistent with hemodynamics, with a progressive increase in the proportion of CNC-derived VSMCs along the ascending aorta to the aortic arch suggesting that the composition of the VSMCs of distinct lineage influences the intensity of the arterial wall. Indeed, VSMCs of different embryonic origins are phenotypically and functionally distinct, with different proliferation and protein expression patterns in response to cytokines
[30][38]. Stimulation with wound repair factor transforming growth factor-beta 1 (TGF-β1) or platelet-derived growth factor BB (PDGF-BB) significantly enhanced the biosynthesis of ECM components such as Col1A1 in CNC-derived VSMCs and promoted cell proliferation by increasing c-myb expression, but there was no effect on VSMCs of mesodermal origin, suggesting that embryonic lineage is a major source of aortic VSMC heterogeneity
[31][32][33][39,40,41].
3. VSMC Phenotypic Diversity in Hereditary AAs
The susceptibility to aortopathy varies between aortic segments. The ascending aorta is less prone to atherosclerosis than the abdominal aorta, and even thoracoabdominal aortic graft exchange does not alter atherosclerotic propensity, suggesting that the intrinsic nature of the arterial wall influences vulnerability to arterial disease
[34][35][36][42,43,44]. Segment-specific distribution of heterogeneous VSMCs likely contributes significantly to aortic disease characteristics. Further support for this view is the fact that the aortic arch, which is composed of CNC-derived VSMCs, is the segment of the thoracic aorta that is more susceptible to calcification
[37][38][45,46]. As VSMCs of different embryonic origins have heterogeneous patterns in response to environmental cues, genetic mutations that can affect intracellular signaling could also have heterogeneous effects on them. Patients with genetic defects in focal adhesion mediators or components of the TGF-β signaling cascade are predisposed to thoracic aortic aneurysms (TAAs) with manifestations of syndromic TAAs such as Marfan syndrome (MFS), Loeys–Dietz syndrome (LDS), or TAAs alone
[39][40][41][42][43][44][45][46][47][47,48,49,50,51,52,53,54,55]. Importantly, almost all AAs in LDS and MFS patients occur in the aortic root and ascending arteries, and dissections often arise in the ascending aorta (
Figure 1B), suggesting that heterogeneous lineage plasticity of VSMCs determines the pathological characteristics of hereditary aneurysms
[48][49][56,57].
3.1. VSMC Phenotypic Diversity in LDS
LDS is an aneurysm-predisposing disease caused by defects in the canonical positive regulators of TGF-β, which regulate the differentiation and maturation of VSMCs
[50][51][58,59]. Heterozygous loss-of-function mutations in genes encoding multiple components of the TGF-β signaling pathway, including the ligands (TGFB2/3), transforming growth factor-β receptor types I and II (TGFBR1 and TGFBR2) and downstream effectors (SMAD2/3), cause abnormalities in the function of VSMCs (
Figure 1C).
Approximately 20–25% of patients with LDS have mutations in the
TGFBR1 genes
[52][60]. The embryonic background of the VSMCs determines the effect of this genetic defect. SHF-derived VSMCs, but not CNC-derived VSMCs, exhibited impaired Smad2/3 activation, increased angiotensin II type 1 receptor (Agtr1a), and TGF-β ligands expression in the LDS model carrying heterozygous Tgfbr1 inactivation
[53][61]. A human-induced pluripotent stem cell (hiPSC) model derived from an LDS patient family with the
TGFBR1A230T variant also detected disruption of SMAD3 and AKT activation and significantly reduced contractile transcript and protein levels in SHF-derived VSMCs, but not in CNC-derived VSMCs
[54][62]. Single-cell transcriptomic data revealed molecular similarities between SHF-derived VSMCs with the
TGFBR1A230T variant and SHF-derived VSMCs with a loss-of-function SMAD3 mutation. Additionally, pharmacological activation of the intracellular SMAD2/3 signaling cascade can rescue contractile gene expression and contractile function in TGFBR1 mutant SHF-derived VSMCs
[54][62].
Approximately 55–60% of LDS patients carry
Tgfbr2 mutations
[52][60]. The effect of Tgfbr2-mediated signaling activation on VSMC gene expression depends on the location of VSMCs in the aorta
[55][68]. Smooth muscle specific
Tgfbr2 deficiency significantly enhances the progression of ascending but not abdominal aortic pathology, including increased intramural hemorrhage, medial thinning, and epicardial thickening, and damage was more pronounced outside of the media layer than inside
[56][57][69,70]. Although rigorous experimental evidence such as lineage-stratified sc-RNAseq and lineage tracing is required, it is speculated that VSMCs of different embryonic origins respond heterogeneously to Tgfbr2 deficiency. Whether
Tgfbr2 exerts a facilitative or inhibitory effect on the differentiation of neural crest cells (NCCs) into VSMCs remains unclear. TGF-β signaling is involved in NCCs migration and differentiation and germline knockout of
Tgfbr2 in VSMCs leads to extensive and lethal cardiovascular malformation in mouse embryos
[55][58][59][60][68,71,72,73].
Tgfbr2 deficiency was found to hinder NCC-to-VSMC differentiation, resulting in a deficiency of CNC-derived VSMCs in the aorta
[60][61][73,74]. However, recent studies have shown that
Tgfbr2 deficiency does not impede the VSMC fate of NCCs and may even lead to premature differentiation of NCCs into VSMCs
[55][62][63][68,75,76]. Smad2 is confirmed as an essential regulator in progenitor-specific VSMC development and physiological differences between CNC- and mesoderm-derived VSMCs
[59][64][65][72,77,78]. Although in vitro studies found Smad2 functionally intact in
Tgfbr2-deficient VSMCs, animal models demonstrated reduced p-Smad2 levels
[57][64][66][70,77,79]. Embryonic
Smad2 knockdown in NCCs reduces the number of CNC-derived VSMCs and produces a damaged, fragmented elastin lamina in the media of the ascending aorta
[57][64][67][70,77,80]. Strikingly, postnatal knockdown of Tgfbr2 in VSMCs, while reducing Tgfb signaling, has little effect on contractile gene expression levels and contractile function in VSMCs, but rapidly induces elastin fragmentation and ascending thoracic aorta disruption (ATAD)
[57][63][70,76].
3.2. VSMC Phenotypic Diversity in MFS
FBN1 gene mutations predispose Marfan syndrome (MFS) patients to TAA at a young age and premature death from catastrophic aneurysm rupture or dissection
[48][56]. The
FBN1 gene does not directly regulate the contraction-related phenotype of VSMC, but its coding product fibrillin-1 is an important component of the ECM and acts to mediate the attachment of VSMCs to elastin laminae
[68][89]. Fibrillin-1, the encoded product of FBN1, is an important component of the ECM that mediates the attachment of VSMCs to elastin laminae, the loss of which would disrupt mechanotransduction and result in detached VSMCs with altered morphology, reduced contractile signatures, and upregulation of several ECM elements and elastolysis mediators indicating cellular plasticity. (
Figure 1C)
[69][90].
MFS VSMCs derived from iPSCs showed a lineage-specific protein profile. Compared to CNC-derived VSMCs, SHF-derived VSMCs have a proteome with less VSMC identity (
TAGLN), but increased levels of focal adhesion components (integrin αV, fibronectin) and ECM remodeling molecules (collagen type 1, MMP2)
[70][91]. As a novel protein marker associated with MFS aneurysms, uPARAP, an endocytic receptor responsible for receptor-mediated internalization of collagen for degradation, was specifically downregulated in SHF-derived VSMCs
[70][71][91,92]. Recent work has shown that uPARAP also acts as an endocytic receptor for thrombospondin-1 (TSP-1), which has been reported to promote AAs by increasing aortic inflammation and TGF-β activity
[72][73][74][75][76][93,94,95,96,97]. Single-cell transcriptomics provides evidence for lineage-specific VSMC plasticity in MFS aortic aneurysms. Albert and colleagues identified a group of modulated VSMCs (modVSMCs) in aortic aneurysm tissue from
Fbn1C1041G/+ mice with a transcriptome similar to that of VSMC-lineage-derived fibromyocytes in atheroma
[77][98]. The combination of lineage tracing and scRNA-seq uncovered that SHF-derived VSMCs and CNC-derived VSMCs contribute equally to modVSMCs
[78][99].
4. VSMC Phenotypic Diversity in Non-Hereditary AAs
Apart from a few that can be explained by causative genetic mutations, most sporadic patients do not have genetic defects and these AAs are called non-hereditary AAs
[79][80][81][103,104,105]. The etiology of non-hereditary AAs is still enigmatic and previous studies have shown that risk factors for non-hereditary AAs, such as hypertension, aging, and hyperlipidemia, transform contractile VSMCs into synthetic VSMCs
[81][82][83][84][85][86][105,106,107,108,109,110]. However, the definition of synthetic VSMCs is ambiguous and recent studies demonstrate that they are actually conglomerates of several VSMC phenotypes with different functions (
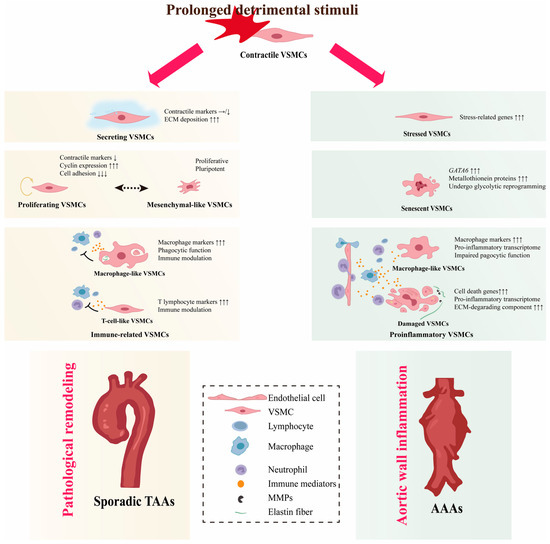
Figure 2).
Figure 2. VSMC phenotypic diversity in non-hereditary AAs. VSMCs transform into different phenotypes and show a high degree of plasticity when exposed to detrimental stimuli. In the thoracic aorta, secreting VSMCs cause deposition of ECM components. Proliferative VSMCs undergo active proliferation and immune-related VSMCs will exert immune clearance and immunomodulatory effects. In the long term, these non-contractile phenotypes can result in pathological remodeling and, ultimately, to TAAs. In the abdominal aorta, stressed VSMCs are standing at the at the crossroads of necrosis/apoptosis or compensation. Pro-inflammatory VSMCs maintain and exacerbate vessel wall inflammation, eventually exhausting proliferative VSMCs and triggering a senescent phenotype, which leads to VSMC depletion and disruption of aortic wall integrity, triggering AAAs. Arrows indicates the gene expression changes: the number of arrows indicates the magnitude of expression change, and the direction of arrows indicates the trend, upward represent upregulation, horizontal indicate stable expression levels, and downward indicate downregulation1.
4.1. VSMC Diversity in TAAs
4.1.1. Secreting VSMCs
Although investigators annotate VSMCs differently, several studies have found a subcluster of VSMCs positioned between VSMCs and fibroblasts in both UMAP and tSNE projections
[87][88][89][90][111,112,113,119]. It maintains or moderately reduces the expression of contraction-related genes, but upregulates genes enriched in cell adhesion and ECM remodeling
[15][16][15,16]. This subcluster is referred to as fibromyocytes or secreting VSMCs due to their properties of both contractile VSMCs and fibroblasts. Unlike “synthetic” VSMCs, secreting VSMCs do not activate cyclin and pro-inflammatory gene expression. Although rare in healthy aortic walls, secreting VSMCs significantly increased their proportion in aneurysmal aorta, further reducing VSMC identity markers such as
ACTA2 and
MYH11 and expressing ECM genes such as
SERPINE1 and
FN1 [90][119].
VCAN, a gene encoding Versican, a chondroitin sulfate proteoglycan composing the ECM, was identified as a marker of secreting VSMCs. Trajectory analysis of VCAN
+ VSMCs further supports the phenotypic transition from contractile VSMCs to fibroblasts
[89][91][113,120]. It should be noted that the transition to secreting VSMCs is common in several aortic diseases. In atheroma, VSMC-traced scRNA-seq confirmed that the fibromyocytes are derived from contractile VSMCs, and they can be clustered with modVSMCs in MFS
[77][92][93][98,121,122].
4.1.2. Proliferating VSMCs
VSMC proliferation is a hallmark of occlusive aortic diseases such as restenosis after angioplasty or stenting
[94][124]. Upon injury to the vascular wall, the quiescent, differentiated VSMCs have been reported to be activated to a proliferative phenotype at low frequencies and such oligoclonal contribution of VSMCs is a feature of obstructive arterial disease
[95][96][97][98][125,126,127,128]. Fate mapping analyses in TAA models induced by disruption of the TGF-β pathway also revealed oligoclonal expanding of the VSMCs
[99][100][115,116]. Enhanced VSMC proliferation was demonstrated in sporadic TAAs
[87][88][101][111,112,114]. In human TAA samples, sc-RNAseq identified a subpopulation of VSMCs characterized by high levels of cyclin expression and low levels of cell adhesion, which is highly suggestive of ongoing cell proliferation and was annotated as proliferating VSMCs
[87][111]. Despite expressing some canonical synthetic marker genes such as
MGP,
TPM4, and
MYH10, the proliferating VSMCs do not upregulate ECM and inflammatory genes, which is different from synthetic VSMCs
[87][111].
4.1.3. Immune-Related VSMCs
In the context of atherosclerosis, VSMCs have been shown to have the potential to switch to monocyte/macrophage lineages, implicating them in the modulation of inflammation in the vascular wall
[98][102][103][104][128,129,130,131]. Although medial degeneration has been confirmed as the most common pathological manifestation of TAAs, aortitis, and atherosclerosis remain the main histopathological substrates in 1/3 of TAA patients
[105][106][132,133]. The prevalence of inflammation-related histologic changes underscores the pivotal role of the inflammatory response in the pathophysiology of TAAs. VSMCs that exhibit similar characteristics of monocyte/macrophage and T-lymphocyte are identified in TAAs and referred to as immune-related VSMCs
[87][88][107][111,112,117]. Additionally, an intermediate state, which expresses interferon-induced genes, has the potency to switch to T-cell-like VSMCs or macrophage-like VSMCs, suggesting that VSMCs are activated by inflammation
[87][111]. Bioinformatics analysis revealed that immune-related VSMCs exert immunomodulatory functions through rich and strong interactions with immune cells
[88][112]. T-cell-like VSMCs recruit immune cells mainly through the CXCL12-CXCR4 pair. Macrophage-like VSMCs in TAAs can communicate with all infiltrating immune cells through PTN and galectin, which are related to the suppressive immune microenvironment in malignancies
[108][109][110][134,135,136]. The immunosuppressive function of macrophage-like VSMCs may explain why the inflammatory response in Ang II-induced TAAs is not as strong as in AAAs.
4.2. VSMC Diversity in AAAs
4.2.1. Proinflammatory VSMCs
AAA is considered to be a chronic inflammatory disease. Pro-inflammatory VSMCs defined by their function to promote aortic wall inflammation are identified. They have a proinflammatory transcriptome characterized by the expression of proinflammatory cytokines (IL-1ß, IL-6, CCL2, CCL5, and TNFα), chemokines (CXCL10 and ICAM1), and high expression of ECM-degrading components such as MMP2/9. Aortic wall inflammation can damage VSMCs, activate the expression of inflammation-related genes through innate immune pathways, and further promote vascular wall inflammation, forming a vicious cycle
[111][112][113][139,140,141]. VSMCs expressing macrophage markers are also present in AAAs.
[29][114][27,118].
4.2.2. Senescent VSMCs
Chronic inflammation of the vessel wall is bound to cause the death of VSMCs, and the depletion of contractile VSMCs is a hallmark of AAAs. The scRNA-seq analyses failed to identify any proliferating VSMCs in the AAA specimens, indicating the failure of the VSMCs to activate into a proliferative phenotype. Nevertheless, a group of VSMCs that specifically express high levels of GATA6 and undergo glycolytic reprogramming has been identified
[114][118]. GATA6 plays a multifaceted role in regulating VSMC phenotype. GATA6 has been implicated in the differentiation of VSMCs, as studies have shown that specific knockout of GATA6 in VSMCs can activate the expression of synthetic markers
[115][116][147,148].
4.2.3. Stressed VSMCs
Stress events such as oxidative stress, mitochondrial dysfunction, and unfolded protein response have been shown to alter the VSMC phenotype toward pro-inflammation and proliferation
[117][118][119][120][153,154,155,156]. A group of VSMCs with transcriptomic features similar to contractile VSMCs, except a characteristically high expression of stress-related genes (
FOS,
ATF3,
JUN, and
HSPB8), were identified in TAAs and annotated as stressed VSMCs
[87][111]. VSMCs with contractile VSMC transcriptome and high
FOS,
Jun,
Klf2, and
ATF3 expression were also identified in elastase-induced AAAs
[114][118]. Due to the relatively high expression of genes associated with proliferation, they were annotated as proliferative VSMCs by the other. However, the actual proliferative capacity of this group of cells is uncertain because they express a high level of mitogen-activated protein kinase phosphatase-1 (Dusp1), which inhibits proliferation by suppressing MAPK activity.
5. Role of VSMC Phenotypic Diversity in AAs
In clinical samples from patients with AAs, changes in VSMC phenotype affect hemostasis of the arterial wall, and VSMC plasticity has been correlated with disease severity. Such studies, however, represent only the final stage of the disease. Thus, whether VSMC heterogeneity and plasticity play a causative or compensatory role in AAs remains controversial. Animal models are tools for studying VSMC pathogenesis and phenotypic disruption of VSMCs can be inferred to cause hereditary aortic aneurysms from genetically engineered models
[54][121][122][62,66,157]. VSMCs of different lineages respond differently to the same genetic mutation, and SHF-derived VSMCs are more phenotypically vulnerable than CNC-derived VSMCs.
In contrast to hereditary AA, sporadic AA is a multifactorial disease that poses difficulties in the construction of animal models. At present, no animal model perfectly mimics human non-genetic AAs
[123][124][125][164,165,166]. Results from non-hereditary models of AA support to some extent, the compensatory role of VSMC phenotypic transformation. To induce AA in WT mice, a minimum of two risk factors as well as sufficient challenge time are required. Challenging the aorta with a single risk factor such as Ang II infusion, hypertension, or hypercholesterolemia alone can elicit phenotypic changes of VSMCs but is not insufficient to induce AAs
[81][82][83][84][85][86][105,106,107,108,109,110]. Extreme agents such as calcium chloride and elastase, which directly destroy the aortic wall, do induce AAs
[126][127][128][167,168,169].
Despite the potential protective effects of the phenotypic alteration of VSMCs, the development and progression of sporadic AAs are indicative of maladaptation. The scholars propose that the sustained presence of noxious stimuli causes a prolonged phenotypic transformation of VSMCs, which causes pathological remodeling of the aortic wall and eventually leads to non-hereditary AAs. Proteoglycan and collagen accumulation prevent aortic dissection and rupture, however, overaccumulation of proteoglycans and collagen results in stiffening of the aortic wall which disrupts the phenotypic change of VSMCs
[129][130][171,172]. Elevated ECM stiffness has been reported to polarize contractile VSMCs toward proinflammatory phenotypes through mechanotransduction
[131][132][133][134][173,174,175,176].