Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Marion Müller.
Molecular processes underlying right ventricular (RV) dysfunction (RVD) and right heart failure (RHF) need to be understood to develop tailored therapies for the abatement of mortality of a growing patient population. Today, the armament to combat RHF is poor, despite the advancing identification of pathomechanistic processes. Mitochondrial dysfunction implying diminished energy yield, the enhanced release of reactive oxygen species, and inefficient substrate metabolism emerges as a potentially significant cardiomyocyte subcellular protagonist in RHF development. Dependent on the course of the disease, mitochondrial biogenesis, substrate utilization, redox balance, and oxidative phosphorylation are affected.
- right heart failure
- mitochondria
- oxidative stress
1. Introduction
Right ventricular dysfunction (RVD) is an independent determinant of mortality in several cardiovascular disorders of right ventricular (RV) pressure or volume overload such as pulmonary arterial hypertension (PAH), congenital heart disease, left heart failure (LHF), or tricuspid valve regurgitation (TR) [1]. Whereas the current understanding of and treatment options for LHF have continuously improved during recent years, no therapeutic options are available in today’s clinical routine that specifically target the right heart [2,3,4][2][3][4]. This implies that significant interventricular differences exist, which are responsible for unsuccessful pharmacological treatment. Besides differences in morphology and hemodynamics, distinct cellular and molecular processes and their regulation under pathophysiological conditions may significantly account for individual maladaptive mechanisms of the right and left ventricles [3,5][3][5]. Whereas the principal pathogenic processes in right heart failure (RHF) are the same as in LHF, their importance and order may be diverse. Importantly, structural remodeling may play a minor role in RHF compared to LHF [6,7,8][6][7][8].
Given that the heart is the most metabolically active organ in the body, it is particularly reliant on mitochondrial integrity. Thus, mitochondrial dysfunction is suggested to play a prominent role in heart failure.
2. The Powerhouse of the Heart
Mitochondria are oval organelles around 0.7 to 1 µm in size containing a double membrane structure with transversally oriented cristae formed by infolding of the inner membrane. They are tightly connected and form chain-like structures assembled in mitochondrial clusters. In the cardiomyocyte, mitochondria can be perinuclear, interfibrillar, and subsarcolemmal and possess different proteomic and physiological statuses [9,10][9][10]. Although mitochondria exhibit a dynamic characteristic, they are the source of energy generating the majority of the adenosine triphosphate (ATP) in the heart. The perpetual energy demand in the heart requires a mitochondrial volume of 23 to 32% of the total cardiomyocyte volume. The cardiac mitochondrial volume increases continuously from humans and dogs to rats, hamsters, and mice. Thus, the increased mitochondrial density correlates well with the increased heart rate and oxygen consumption in smaller animals [11]. In mitochondria, energy is generated via oxidative phosphorylation (OXPHOS). This complex process can be described as a sequential passage of electrons from negative (nicotinamide adenine dinucleotide, NADH, and flavin adenine dinucleotide, FADH2) to positive (molecular oxygen O2) redox potentials down the electron transport chain (ETC) in aerobic respiration. OXPHOS normalized to the number of mitochondria reflects the generation of energy in terms of ATP and is defined as a mitochondrial function. The ETC consists of four protein complexes located at the inner mitochondrial membrane. The main energy conversion reactions take place at complex I (NADH dehydrogenase), complex III (cytochrome c reductase), and complex IV (cytochrome c oxidase), which use the energy of the stepwise electron transfer for active pumping of hydrogen ions (H+) out of the mitochondrial matrix into the intermembrane space. Thereby, an electrochemical gradient is established across the inner mitochondrial membrane. The translocation of H+ back into the mitochondrial matrix through the F1FO ATPase (ATP synthase complex) is coupled to the phosphorylation of ADP to generate ATP. The reduced coenzymes NADH and FADH2, the electron donors for the ETC, are produced in the tricarboxylic acid (TCA) cycle within the mitochondrial matrix. One enzyme of the TCA cycle, succinate dehydrogenase or complex II, is part of the ETC located in the inner mitochondrial membrane. Mitochondrial function can be determined by respirometry, which measures the O2 consumption rate in permeabilized muscle fibers, cardiac tissue samples, or isolated mitochondria after the addition of saturating concentrations of metabolic substrates and ADP. Of note, even by using living cells, the information is limited to the general function of the ETC protein complexes, their connectivity and coupling to the ATPase, and the lack of data on the availability of NADH, FADH2, ADP, or O2.3. Redox-Optimized ROS Balance in the Heart
The ETC releases the superoxide radical (O2•−), a reactive oxygen species (ROS), even under physiological conditions, given that a small percentage of electrons prematurely reduce O2 at complex I and complex III. This production of mitochondrial ROS (mitoROS) is balanced by an antioxidant system located in the mitochondrial matrix [12]. The manganese-dependent superoxide dismutase (mitochondrial isoform: SOD2) converts O2•− to O2 and hydrogen peroxide (H2O2), which is then further reduced to water by glutathione peroxidase (GPX) and peroxiredoxin (mitochondrial isoforms: PRX3 and PRX5). For the regeneration of GPX and PRX, the pyridine nucleotide NADPH is needed, whose reduction in mitochondria depends on TCA cycle activity. Therefore, TCA cycle enzyme integrity is essential for ATP production and the regeneration of antioxidative pathways. Pathological conditions can lead to dysfunction of the TCA cycle and OXPHOS in mitochondria, which effects ATP production and the generation of mitoROS due to an altered redox balance (NAD+/NADH, FAD/FADH2, NADP+/NADPH ratios). Under physiological conditions, the emission of mitoROS in the ETC is low, allowing the antioxidant system to balance ROS levels sufficiently with adequate NADPH availability for the regeneration of the antioxidant enzyme systems. However, under pathophysiological conditions, the ETC becomes uncoupled, resulting in a decreased membrane potential and less ATP generation. An extreme oxidized state, probably due to altered metabolic flows and less TCA cycle activity and therefore lower restoration of reduced coenzymes NADH and FADH2, depletes H2O2 scavenging capacity and the net ROS emission is enhanced despite eventually lower ROS production (Figure 1).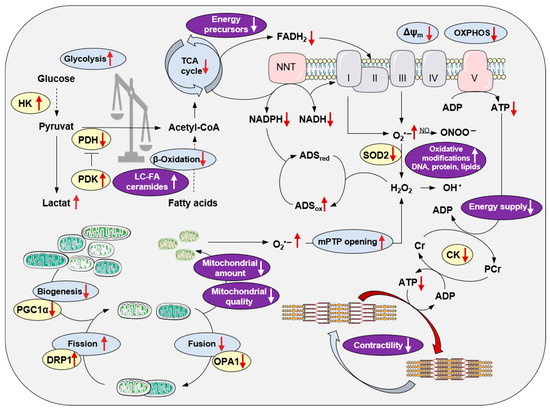
Figure 1. Modifications of cardiac mitochondrial integrity in right heart failure. Principal processes of energy metabolism, the mitochondrial electron transport chain, oxidative phosphorylation, redox regulations, mitochondrial biogenesis and dynamics, and their impact on the sarcomere are depicted. Red arrows indicate alterations in right heart failure. The purple color indicates the pathological consequences of mitochondrial disintegration. For better clarity, the reaction equations are simplified. Abbreviations: ADS, antioxidative defense system; Cr, creatine; CK, creatine kinase; DRP1, dynamin-related protein1; HK, hexokinase; LC-FA, long-chain fatty acids; mPTP, mitochondrial permeability transition pore; NNT, nicotinamide nucleotide transhydrogenase; OPA1, optic atrophy 1; OXPHOS, oxidative phosphorylation; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PGC1α, peroxisome proliferator-activated receptor γ coactivator 1α; SOD, superoxide dismutase; TCA, tricarboxylic acid.
4. Mitochondrial Integrity Is Impaired in Right Heart Failure
In order to assess the significance of mitochondrial function in the development of RHF, the experimental model system needs to be considered critically. As RHF is a leading cause of mortality in PAH patients, many experimental studies have used rodent models of PAH, induced either by injection of the vascular endothelial growth factor (VEGF) inhibitor, Sugen 5416, followed by hypoxia (SuHx) or the injection of monocrotaline (MCT), a pneumotoxic pyrrolizidine alkaloid. Both model systems are characterized by pulmonary arterial (PA) remodeling and constriction with increased PA pressure and pulmonary vascular resistance leading to compensated RV hypertrophy and eventually RHF. In contrast to the PAH rodent model systems, the model of PA banding (PAB) is widely used to study RV remodeling independently of pulmonary vascular disease. Of note, different mouse wild-type strains might react differently to RV pressure overload, either developing compensated RV dysfunction or failure [6], which was also observed by Shults et al. [29][20] in SuHx Sprague Dawley and Fisher rats. It might be obvious that the degree of afterload triggered by the degree of PA stenosis in PAB rodent models is associated with the extent of RV dysfunction and structural remodeling [6,37][6][21]. However, to assess the molecular mechanisms mediating the transition of compensated RV hypertrophy (RVH) and RVD to RHF, it is essential to determine the signs of RHF in experimental rodent models. Signs of congestion reliably reflect the presence of RHF, as presented in experimental studies, i.e., liver weight [31[22][23][24][25],32,33,34], macroscopic signs of a nutmeg-colored liver [6], and ascites. In addition, impaired exercise capacity [34][25] and/or mortality [6,24,29][6][20][26] can reflect RHF. A quantitative parameter to determine signs of RHF might be the morphometric assessment of congestion-modified areas in haematoxylin/eosin-stained liver sections. The extent of hepatic congestion showed a strong positive correlation with right atrial (RA) area and a strong negative correlation with systolic RV function assessed by echocardiography in PAB mice [6]. To understand RHF, it is important to consider that already under physiological conditions, the RV differs from the left ventricle (LV) in loading conditions and morphology and displays fundamental differences on the cellular and molecular levels. There is accumulating evidence that the RV is more susceptible to oxidative stress probably because of a less adaptable antioxidant enzyme system. In healthy rats, Nagendran et al. [38][27] measured a lower mitochondrial membrane potential in RV cardiomyocytes and RV tissue compared to the LV, which might provide evidence for a lower activity of the ETC complexes in RV compared to LV tissue. Furthermore, the membrane potential rose with disease severity in a mouse model of PAH induced by MCT [38][27], indicating adaptation of RV cardiomyocytes to increasing workloads. Interestingly, Phillips et al. detected an identical expression pattern and relative amount of proteins, the same amount of post-translational protein modifications, and an identical amount of mitochondria in RV and LV tissue of healthy rabbits and pigs [39][28]. Given the lower oxygen consumption and lower ATP generation rate [40,41][29][30] of the resting RV, the RV should hold larger metabolic reserve capacities than the LV. This should imply lower mitoROS generation and a less vulnerable RV under an increasing workload. But in contrast to the LV showing enhanced antioxidative capacity under the progression of pathological conditions [22[31][32][33],23,36], the antioxidant enzyme system is unchanged or is even decreased in the RV myocardium of mice after RV pressure overload [6[6][25],34], rats with chronic NO deficiency [36][33], and in human end-stage HF patients secondary to ischemic HF or idiopathic dilated cardiomyopathy [21,22,23][31][32][34]. To detect the status of the antioxidative system in the RV, several preclinical studies have measured SOD expression on mRNA levels [6,34,36][6][25][33] with differentiation between Sod1, an isoform mainly expressed in the cytosol, and Sod2, the mitochondrial isoform. In studies using human RV myocardium, CAT activity was determined [22,23][31][32] in the tissue of end-stage LHF patients. Progressive destruction of the mitochondrial network, which may result in impaired formation of ATP due to decreased OXPHOS under aerobic conditions, is associated with the development of RHF (Figure 1). A decreased amount of mitochondria has been shown in experimental PAH [25,32,42][23][35][36] and RV pressure overload [6,34][6][25] by detecting decreased activity of citrate synthetase (CS), the pace-maker enzyme of the TCA cycle in mitochondria, or decreased amounts of mtDNA. Experimental data could be confirmed in RV tissue samples of congenital heart disease patients divided into two groups of compensated RVH and RHF based on postoperative cardiac magnetic resonance imaging (MRI) [43][37]. While CS and succinate dehydrogenase activities were maintained in RVH, they were significantly decreased in RHF. The mtDNA copy number progressively decreased during the transition from RVH to RHF and inversely correlated with RV systolic pressure [43][37]. In contrast with reduced mitochondrial biogenesis, reflected by decreased Pparagc1a (PGC1α) mRNA levels, reported in SuHx rats [25][35], Karamanlidis et al. showed significantly increased Pparagc1a (PGC1α) mRNA expression in human RVH and again normalized mRNA levels in human RHF [43][37]. This might indicate mitochondrial biogenesis as a highly dynamic, compensatory effect during the progression of RVH/RVD to RHF. The mitochondrial architecture is also influenced by impaired mitochondrial fission and fusion (Figure 1), a process by which mitochondria divide or fuse to maintain their integrity and function [44][38]. Of note, increased mitochondrial fission as well as enhanced fusion are reported in experimental PAH [31,45,46,47][22][39][40][41]. The fission/fusion ratio and thereby mitochondrial biogenesis might also depend on disease severity, as Hwang et al. [34][25] showed significantly increased mitochondrial fusion in compensated RVH upon RV pressure overload by protein expression of mitochondrial dynamin-like GTPase (OPA1). Both OPA1 isoforms were significantly decreased in RHF upon RV pressure overload, while the mitochondrial fission protein, dynamin-related protein (DRP1), was significantly increased in isolated RV mitochondria in RHF upon PAB [34][25]. The same expression pattern, which ultimately leads to an impaired fission/fusion ratio in RVH, was also shown by Wüst et al. [42][36] in MCT rats. After injection of low-dose MCT resulting in compensated RVH, increased fusion and decreased fission were observed, while rats injected with a higher MCT dosage suffered from RHF and showed disrupted fission as well as fusion [42][36]. In accordance, data from transmission electron microscopy (TEM) of RV tissue showed an elevated mitochondrial area and increased mitochondrial size and higher amounts of mitochondrial clusters with a decreased number of mitochondria per cluster in the RV of SuHx rats at early time points. In contrast, at later time points, the mitochondrial area significantly decreased and mitochondrial integrity was disrupted, which was associated with increased ROS generation and reduced energy production in the RV tissue of SuHx rats [29][20]. An impaired mitochondrial ultrastructure was also shown in other PAH rodent models [25[23][35][36],32,42], models of RV pressure overload [25,34][25][35], and in human RV tissue of congenital heart disease patients [43][37].References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731.
- Klinke, A.; Schubert, T.; Muller, M.; Legchenko, E.; Zelt, J.G.E.; Shimauchi, T.; Napp, L.C.; Rothman, A.M.K.; Bonnet, S.; Stewart, D.J.; et al. Emerging therapies for right ventricular dysfunction and failure. Cardiovasc. Diagn. Ther. 2020, 10, 1735–1767.
- Reddy, S.; Bernstein, D. Molecular Mechanisms of Right Ventricular Failure. Circulation 2015, 132, 1734–1742.
- Tadic, M.; Pieske-Kraigher, E.; Cuspidi, C.; Morris, D.A.; Burkhardt, F.; Baudisch, A.; Hassfeld, S.; Tschope, C.; Pieske, B. Right ventricular strain in heart failure: Clinical perspective. Arch. Cardiovasc. Dis. 2017, 110, 562–571.
- Friedberg, M.K.; Redington, A.N. Right versus left ventricular failure: Differences, similarities, and interactions. Circulation 2014, 129, 1033–1044.
- Muller, M.; Bischof, C.; Kapries, T.; Wollnitza, S.; Liechty, C.; Geissen, S.; Schubert, T.; Opacic, D.; Gercek, M.; Fortmeier, V.; et al. Right Heart Failure in Mice Upon Pressure Overload Is Promoted by Mitochondrial Oxidative Stress. JACC Basic Transl. Sci. 2022, 7, 658–677.
- Boehm, M.; Tian, X.; Mao, Y.; Ichimura, K.; Dufva, M.J.; Ali, K.; Dannewitz Prosseda, S.; Shi, Y.; Kuramoto, K.; Reddy, S.; et al. Delineating the molecular and histological events that govern right ventricular recovery using a novel mouse model of pulmonary artery de-banding. Cardiovasc. Res. 2020, 116, 1700–1709.
- Crnkovic, S.; Egemnazarov, B.; Damico, R.; Marsh, L.M.; Nagy, B.M.; Douschan, P.; Atsina, K.; Kolb, T.M.; Mathai, S.C.; Hooper, J.E.; et al. Disconnect between Fibrotic Response and Right Ventricular Dysfunction. Am. J. Respir. Crit. Care Med. 2019, 199, 1550–1560.
- Kuznetsov, A.V.; Usson, Y.; Leverve, X.; Margreiter, R. Subcellular heterogeneity of mitochondrial function and dysfunction: Evidence obtained by confocal imaging. Mol. Cell. Biochem. 2004, 256, 359–365.
- Kurz, F.T.; Aon, M.A.; O’Rourke, B.; Armoundas, A.A. Functional Implications of Cardiac Mitochondria Clustering. In Mitochondrial Dynamics in Cardiovascular Medicine; Springer: Berlin/Heidelberg, Germany, 2017; Volume 982, pp. 1–24.
- Schaper, J.; Meiser, E.; Stammler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391.
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877.
- Kaludercic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332.
- Carnicer, R.; Crabtree, M.J.; Sivakumaran, V.; Casadei, B.; Kass, D.A. Nitric oxide synthases in heart failure. Antioxid. Redox Signal 2013, 18, 1078–1099.
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194.
- Rothfuss, O.; Gasser, T.; Patenge, N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res. 2010, 38, e24.
- Yan, L.J. Analysis of oxidative modification of proteins. Curr. Protoc. Protein Sci. 2009, 14.
- Budde, H.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Herwig, M.; Varatnitskaya, M.; Sieme, M.; Delalat, S.; Sultana, I.; Kolijn, D.; et al. The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts. Antioxidants 2021, 10, 1134.
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8197–8202.
- Shults, N.V.; Kanovka, S.S.; Ten Eyck, J.E.; Rybka, V.; Suzuki, Y.J. Ultrastructural Changes of the Right Ventricular Myocytes in Pulmonary Arterial Hypertension. J. Am. Heart Assoc. 2019, 8, e011227.
- Rain, S.; Andersen, S.; Najafi, A.; Gammelgaard Schultz, J.; da Silva Goncalves Bos, D.; Handoko, M.L.; Bogaard, H.J.; Vonk-Noordegraaf, A.; Andersen, A.; van der Velden, J.; et al. Right Ventricular Myocardial Stiffness in Experimental Pulmonary Arterial Hypertension: Relative Contribution of Fibrosis and Myofibril Stiffness. Circ. Heart Fail 2016, 9, e002636.
- Redout, E.M.; van der Toorn, A.; Zuidwijk, M.J.; van de Kolk, C.W.; van Echteld, C.J.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1038–H1047.
- Power, A.S.; Norman, R.; Jones, T.L.M.; Hickey, A.J.; Ward, M.L. Mitochondrial function remains impaired in the hypertrophied right ventricle of pulmonary hypertensive rats following short duration metoprolol treatment. PLoS ONE 2019, 14, e0214740.
- Zimmer, A.; Teixeira, R.B.; Bonetto, J.H.P.; Bahr, A.C.; Turck, P.; de Castro, A.L.; Campos-Carraro, C.; Visioli, F.; Fernandes-Piedras, T.R.; Casali, K.R.; et al. Role of inflammation, oxidative stress, and autonomic nervous system activation during the development of right and left cardiac remodeling in experimental pulmonary arterial hypertension. Mol. Cell Biochem. 2020, 464, 93–109.
- Hwang, H.V.; Sandeep, N.; Nair, R.V.; Hu, D.Q.; Zhao, M.; Lan, I.S.; Fajardo, G.; Matkovich, S.J.; Bernstein, D.; Reddy, S. Transcriptomic and Functional Analyses of Mitochondrial Dysfunction in Pressure Overload-Induced Right Ventricular Failure. J. Am. Heart Assoc. 2021, 10, e017835.
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960.
- Nagendran, J.; Gurtu, V.; Fu, D.Z.; Dyck, J.R.; Haromy, A.; Ross, D.B.; Rebeyka, I.M.; Michelakis, E.D. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J. Thorac. Cardiovasc. Surg. 2008, 136, 168–178.
- Phillips, D.; Aponte, A.M.; Covian, R.; Neufeld, E.; Yu, Z.X.; Balaban, R.S. Homogenous protein programming in the mammalian left and right ventricle free walls. Physiol. Genom. 2011, 43, 1198–1206.
- Zong, P.; Tune, J.D.; Downey, H.F. Mechanisms of oxygen demand/supply balance in the right ventricle. Exp. Biol. Med. 2005, 230, 507–519.
- Bogaard, H.J.; Abe, K.; Vonk Noordegraaf, A.; Voelkel, N.F. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009, 135, 794–804.
- Borchi, E.; Bargelli, V.; Stillitano, F.; Giordano, C.; Sebastiani, M.; Nassi, P.A.; d’Amati, G.; Cerbai, E.; Nediani, C. Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim. Biophys. Acta 2010, 1802, 331–338.
- Manni, M.E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine Oxidase Is Overactivated in Left and Right Ventricles from Ischemic Hearts: An Intriguing Therapeutic Target. Oxid. Med. Cell Longev. 2016, 2016, 4375418.
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schluter, K.D. Specific Mechanisms Underlying Right Heart Failure: The Missing Upregulation of Superoxide Dismutase-2 and Its Decisive Role in Antioxidative Defense. Antioxid. Redox Signal 2015, 23, 1220–1232.
- Nediani, C.; Borchi, E.; Giordano, C.; Baruzzo, S.; Ponziani, V.; Sebastiani, M.; Nassi, P.; Mugelli, A.; d’Amati, G.; Cerbai, E. NADPH oxidase-dependent redox signaling in human heart failure: Relationship between the left and right ventricle. J. Mol. Cell Cardiol. 2007, 42, 826–834.
- Gomez-Arroyo, J.; Mizuno, S.; Szczepanek, K.; Van Tassell, B.; Natarajan, R.; dos Remedios, C.G.; Drake, J.I.; Farkas, L.; Kraskauskas, D.; Wijesinghe, D.S.; et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ. Heart Fail 2013, 6, 136–144.
- Wust, R.C.; de Vries, H.J.; Wintjes, L.T.; Rodenburg, R.J.; Niessen, H.W.; Stienen, G.J. Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc. Res. 2016, 111, 362–372.
- Karamanlidis, G.; Bautista-Hernandez, V.; Fynn-Thompson, F.; Del Nido, P.; Tian, R. Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ. Heart Fail 2011, 4, 707–713.
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884.
- Tian, L.; Neuber-Hess, M.; Mewburn, J.; Dasgupta, A.; Dunham-Snary, K.; Wu, D.; Chen, K.H.; Hong, Z.; Sharp, W.W.; Kutty, S.; et al. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J. Mol. Med. 2017, 95, 381–393.
- Tian, L.; Potus, F.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Lima, P.; Archer, S.L. Increased Drp1-Mediated Mitochondrial Fission Promotes Proliferation and Collagen Production by Right Ventricular Fibroblasts in Experimental Pulmonary Arterial Hypertension. Front. Physiol. 2018, 9, 828.
- Xiong, P.Y.; Tian, L.; Dunham-Snary, K.J.; Chen, K.H.; Mewburn, J.D.; Neuber-Hess, M.; Martin, A.; Dasgupta, A.; Potus, F.; Archer, S.L. Biventricular Increases in Mitochondrial Fission Mediator (MiD51) and Proglycolytic Pyruvate Kinase (PKM2) Isoform in Experimental Group 2 Pulmonary Hypertension-Novel Mitochondrial Abnormalities. Front. Cardiovasc. Med. 2018, 5, 195.
More