1. Introduction
About 80% of individuals with COVID-19 had reported mild to moderate symptoms, and 5% underwent severe illness
[1][6]. Then, up to 70% of COVID-19 survivors may develop long-term medical consequences in addition to significant morbidity and mortality in the initial few weeks following infection. Long after patients are virus-free, the persistent symptoms from COVID-19 infection can drastically lower the quality of life. The long-term COVID-19 effect or extended COVID-19 is another name for this post-COVID-19 situation
[2][7]. This condition is known as “long COVID”, “long haulers”, or “post-COVID syndrome”
[3][4][5][8,9,10]. In May 2020, the phrase “long COVID” was first used
[6][11].
From various biomarkers there are several choices of biomarkers that can be used in post-COVID-19 inflammatory and coagulation conditions, as follows.
2. D-Dimer
D-dimer is an epitope formed when plasmin breaks down fibrin crosslinks. Several disorders, including thrombosis, DIC, and inflammation, raise D-dimer levels. By stimulating the release of various inflammatory cytokines from neutrophils and monocytes, D-dimer can aid in the development of inflammation
[7][49]. Numerous viral infections have been said to cause thrombosis and bleeding as side effects. The development of microvascular thrombus in numerous organs might worsen DIC as a result of an excessive reaction to infection
[8][50]. Tang et al. (2020) found coagulopathy in COVID-19 pneumopathy at an advanced stage with elevated levels of D-dimer and fibrinogen. Due to plasmin-associated hyperactive fibrinolysis and the plasminogen/plasmin system’s involvement in COVID-19 illness, highly elevated D-dimer is produced
[9][19]. Concerns about the cohabitation of venous thromboembolism aggravating ventilation–perfusion mismatch have been raised by the discovery of higher d-dimer levels in COVID-19 patients, and numerous investigations have demonstrated the prevalence of pulmonary embolism. Clinicians would be hesitant to prescribe endogenous anticoagulants to everyone, nevertheless, due to the elevated risk of bleeding and discouragement associated with earlier unsuccessful trials of them in sepsis. It is evident that the effects of coagulation activation go beyond clotting and that the interaction between coagulation and inflammation can profoundly affect disease progression and result in unfavorable outcomes beyond the prevention and management of venous thromboembolism
[10][42].
Various studies state an increase in D-dimer with post-COVID-19 conditions. Fazio et al. (2020) stated after complete clinical recovery from COVID-19, restoration of well-being, and normalization of a molecular swab, 20% of patients had significantly increased D-dimer levels following full clinical recovery of COVID-19, restoration of health, and normalization of a molecular swab. These levels gradually decreased after roughly two weeks of preventive enoxaparin medication
[11][51]. As a result, it is possible that continued elevation of D-dimer levels after a patient has recovered clinically will serve as a warning to identify the processes underlying the frequent long-term effects of SARS-CoV-2 infection. Furthermore, another study by Lehmann et al. (2021) reported that after a median of 3 months following COVID-19, a persistent D-dimer increase was seen in 15% of the individuals who had recovered from COVID-19. These individuals had a more severe COVID-19 infection before
[12][52].
Additionally, several participants who received mRNA injections from Moderna and Pfizer showed an increase in D-dimer, suggesting that they were at risk of clotting. The clinical spectrum ranged from no symptoms to severe, necessitating hospitalization, and D-dimer levels were tested the day before immunization and after 5–7 days
[13][53].
D-dimer may, therefore, be thought of as an easy, trustworthy, and affordable test to monitor and follow COVID-19 patients who need to continue taking low molecular weight heparin or other blood thinners after achieving clinical recovery. But, due to the low specificity of the D-dimer test, there has been an extensive search for alternative or additional biomarkers for specific diagnosis of specific clinical coagulation conditions related D-dimer evaluation
[14][54].
3. Interleukin-6 (IL-6)
Interleukin-6, or IL-6, is a pleiotropic proinflammatory cytokine that is produced by a variety of cell types, including lymphocytes, monocytes, and fibroblasts. Bronchial epithelial cell-dependent production of IL-6 is induced by SARS-CoV-2 infection. A major finding in COVID-19 patients is cytokine dysregulation. Recent research in severely ill COVID-19 patients reveals that the excessive and uncontrolled release of proinflammatory cytokines, known as “Cytokine storm syndrome”, in the body of an infected patient during infection is directly associated with the deregulation of the immune response to the virus
[15][55].
Significant pro-inflammatory properties are exhibited by IL-6, which plays an important role in cytokine storm syndrome, as elevated serum IL-6 levels correlate with respiratory failure and ARDS, which are mediated by two major signaling pathways: cis and trans
[16][56]. In the cis signaling pathway, when IL-6 interacts with membrane-bound IL-6 receptors (IL-6R) and gp130, downstream Janus kinases (JAKs) and also signal transducer and activator of transcription 3 (STAT3) are activated. Activation of this signaling cascade affects the innate immune system (macrophages, neutrophils, and natural killer cells) and acquired immune system (B and T cells), both of which contribute to CRS in a variety of ways. In trans-signaling, circulating IL-6 binds to the soluble IL-6 receptor (sIL-6R) and, in the majority of somatic cell types, forms a complex with the gp130 dimer. Endothelial cells and other cells that do not express mIL-6R become activated as a consequence of the IL-6-sIL-6R-JAK-STAT3 signaling. It substantially worsens the “cytokine storm” through the release of vascular endothelial growth factor (VEGF), monocyte chemoattractant protein-1 (MCP-1), more IL-6 and IL-8, and decreased endothelial E-cadherin expression. VEGF secretion and decreased E-cadherin expression, both of which increase vascular permeability and leakage, play a role in the etiology of pulmonary dysfunction in ARDS
[16][56].
Enhanced blood levels of IL-6, C-reactive protein (CRP), D-dimer, and ferritin are indicators of increased cytokine release and may be linked to systemic inflammation and hypoxemic respiratory failure caused by COVID-19. The length and/or severity of COVID-19 may be lessened by modifying IL-6 levels or IL-6’s effects
[17][57].
IL-6 levels are markedly increased and linked to poor clinical outcomes in COVID-19 patients. High-quality trials of intervention in this area are urgently needed. Inhibition of IL-6 may be a novel target for therapies for the control of dysregulated host responses in patients with COVID-19
[18][58]. Evidence showing a direct correlation between circulating IL-6 levels and the severity of COVID-19 infection was reported. In order to control inflammatory and immune reactions, cytokines are essential. Due to its pleiotropic effects, IL-6 stands out as being particularly significant among them. On the other hand, prior research has shown that individuals with respiratory dysfunction have elevated IL-6 levels
[19][20][59,60], suggesting a possible common mechanism of cytokine-mediated lung damage brought on by COVID-9 infection. The highly pathogenic SARS-CoV-2 also appears to be linked to rapid virus replication and a propensity to infect the lower respiratory tract, leading to an increased response of IL-6-induced acute respiratory distress. As a result, their findings imply that repeated measurements of circulating IL-6 levels may be crucial for detecting illness development in COVID-19-infected individuals
[19][59].
4. Hepatocyte Growth Factor (HGF)
Hepatocyte Growth Factor (HGF) is produced by mesenchymal cells and functions paracrine in epithelial and endothelial cells as a mitogen, motogen, and morphogen. Tyrosine kinase activation is brought on by HGF’s binding to the c-Met receptors on epithelial cells. Blood coagulation serine proteases, such as tissue-type plasminogen activator (tPA), urokinase, factor XIIa, and factor X, cleave the inactive single-chain precursor (pro-HGF) between Arg and Val to transform it into active HGF. The HGF transforming potency of factor XIIa is 1000 times larger than that of tPA or urokinase, making it the most effective activator of them all. Because thrombin transforms the zymogen of the HGF activator into the active form, this conversion appears to be connected to blood coagulation. Urokinase-type (uPA) and tissue-type (tPA) plasminogen activators, coagulation factor XIIa (the most potent activator), and a serine protease homologous to coagulation factor XII, known as an activator of HGF, which is a substrate of thrombin, are the four known plasminogen family proteases that activate pro-HGF. In addition to encouraging the release of active HGF from granulocytes into the bloodstream, coagulation factor Xa also cleaves HGF in the chain, producing an N-terminal fragment with diminished biological activity but the ability to still bind receptors
[21][36]. Therefore, one can draw a conclusion that an activated blood clotting system raises blood levels of active HGF through a variety of its effectors.
Circulating pro-HGF is in equilibrium with that embedded in the dense tissue extracellular matrix. Here, pro-HGF is mostly secreted by stromal cells and stored by coupling with heparan sulfate in the proximity of target cells, which include endothelial and most epithelial cell types. Vascular damage leads to exposure of pro-HGF-bound matrix to blood coagulation enzymes, HGF formation, and stimulation of MET-expressing cells. Interestingly, HGF is not only a downstream effector of blood coagulation but also an activator, as it increases the transcription of genes controlling hemostasis, such as plasminogen activator inhibitors (PAI-1) and cyclooxygenase 2 (COX-2). PAI-1 on fibrinolysis, while COX-2 catalyzes steps in the synthesis of prostaglandins and prostacyclin, which control platelet function
[22][46].
The study by Mosevoll et al., 2015, which examined the numerous soluble inflammatory-related mediators in plasma samples and examined significantly different plasma biomarker profiles between patients with DVT, patients treated for suspected thrombosis but without DVT, and healthy individuals, discussed the role of HGF in coagulation. HGF was one of the mediators the study found to be significantly different between DVT patients and those without thrombus
[14][54]. Another previous study by Perreau et al. (2021) regarding HGF in COVID-19 provides insight into the early pathophysiological events associated with severe COVID-19. Elevated serum HGF concentrations early in symptomatic infection and their association with ICU admission are possible indicators of an ongoing severe respiratory syndrome associated with interstitial pneumonia. HGF upregulation is a physiological counter-regulatory immune response of the host to reduce inflammation, limit lung tissue injury, and promote tissue repair. Consistent with this view, more than 90% of non-ICU patients with moderate respiratory syndrome have low HGF levels, thus identifying HGF as one of the critical pathogenic biomarkers for disease severity and the best predictor of the risk of ICU admission and death in COVID-19 sufferers. Nonetheless, the role of HGF as a biomarker for coagulation in survivors of COVID-19 is unknown
[23][61]. A study by Tamayo-Velasco et al. (2021) explains HGF as a biomarker which substantially related to severe/critical COVID-19 patients at hospital admission and is hence an excellent indicator of poor prognosis and also used as a mortality biomarker
[24][62].
When inflammatory cytokines, such as IL-1, which are potent inducers of pro-HGF transcription, are released in infected patients, stromal cells in tissues may overexpress pro-HGF. HGF and plasma IL-6 levels were shown to correlate in DIC patients
[25][47]. It is interesting to note that the association held true for cancer patients as well as for everyone else who is currently dealing with open DIC. It would be intriguing to look into whether HGF levels in the cancer group were associated with DIC scores, at least in those patients who had high levels of inflammatory cytokines
[21][36]. The absence of a connection between HGF level and DIC severity across the entire cancer cohort may be a result of the complex interplay between blood clotting, tumor tissue, and HGF. Tumor cells can actually: (a) produce HGF; (b) secrete PAI-1, which prevents plasminogen activator from cleaving HGF into its component proteins; (c) interfere with blood coagulation in a number of ways, including tissue factor expression and release via microvesicles; production of molecules (like mucin) that control platelet aggregation; and Secretion of cytokines that activate the endothelium. Levels of pro-HGF and active HGF are probably unrelated to coagulopathy’s supportive mechanisms in this complicated situation
[22][46]. Additionally, it was revealed in a study by Chung et al. (2010) that HGF influences blood clotting and promotes the growth of DIC
[25][47].
HGF, also known as the scatter factor, plays a crucial role in various biological processes, including the blood clotting system. While HGF is not directly involved in blood clotting itself, it influences several factors and pathways that regulate the process. It primarily acts on hepatocytes, endothelial cells, and platelets to modulate their functions and interactions, which indirectly affects blood clotting
[26][27][63,64].
4.1. Endothelial Cell Function
HGF promotes endothelial cell proliferation and migration, leading to the formation of new blood vessels. This angiogenic effect is essential during wound healing and tissue repair, where the development of new blood vessels is crucial for delivering oxygen and nutrients. Proper vascularization is essential for the blood clotting process, as it ensures an adequate blood supply to the injured area
[22][46]. According to reports, COVID-19 may be impacted by endothelial dysfunction and a loss of endothelial barrier function. Heparanase, an endothelial glycocalyx-degrading enzyme, is widely known to play a role in vascular leakage and inflammation. Heparanase is inhibited by low molecular weight (LMW) heparins
[28][65]. Heparinases are enzymes that selectively split the chains of heparin and heparan sulfate by cleaving the glycosidic bond forming between hexosamines and uronic acids, producing disaccharide and oligosaccharide products. Heparin is well known as an anti-coagulant medication but is also implicated in biological processes, e.g., inflammation, cancer, angiogenesis, and viral and bacterial infections. Heparin and heparan sulfate are gaining attention due to their potential as therapeutics and may play a role in the COVID-19 infection, according to recent research. LMW heparin, which has taken the role of heparin in various clinical uses, is produced by the industrial exploitation of heparinases’ capacity to cleave heparin chains selectively. Heparinases are also used to analyze the structural properties of heparin and heparan sulfate, neutralize heparin in blood, and remove heparin’s inhibitory effects on other enzymes
[29][66].
4.2. Platelet Activation and Aggregation
HGF stimulates platelet activation, resulting in the release of various factors from platelet granules. These factors, such as ADP, thromboxane A2, and serotonin, contribute to platelet aggregation and the formation of a stable blood clot. HGF also enhances platelet adhesion to the endothelial cells lining blood vessels, which is a critical step in initiating clot formation
[26][63].
4.3. Hepatocyte Production of Coagulation Factors
The liver is the only primary site of production for many coagulation factors, including Factor Xa. HGF stimulates the growth and regeneration of hepatocytes, which ensures an adequate supply of coagulation factors for the blood clotting cascade. Disruptions in HGF signaling can potentially impact the synthesis and availability of these factors, leading to deficiencies and impairments in the blood clotting system
[30][67].
Recent failures of clinical trials targeting Factor Xa inhibitors have highlighted the need for alternative drug targets or advancements. Factor Xa inhibitors are anticoagulant medications that work by blocking the activity of Factor Xa, a key enzyme involved in the blood clotting cascade. While these inhibitors have shown efficacy in preventing and treating thrombotic disorders, some patients experienced adverse events, including bleeding complications. Identifying alternative drug targets or inhibitors is crucial for advancing anticoagulant therapies. Potential strategies could involve targeting other enzymes involved in the coagulation cascade, such as thrombin or Factor VIIa, or developing novel approaches to modulate platelet activation and aggregation. Additionally, advances in understanding the signaling pathways and interactions involving HGF could offer new insights for therapeutic interventions in the blood clotting system
[31][32][68,69]. Also, further research is needed to explore alternative drug targets and develop safer and more effective anticoagulant therapies, ensuring a balance between preventing thrombosis and minimizing the risk of bleeding complications.
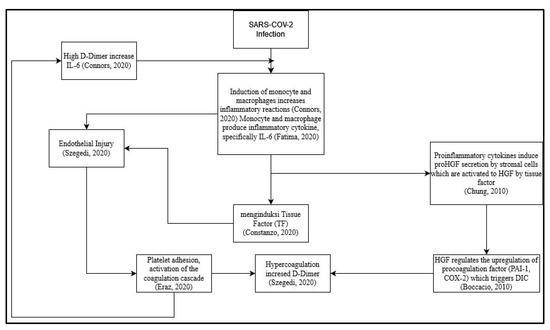
However, until now, the role of HGF in coagulation risk in post-COVID-19 patients is unknown, and how it correlates with D-dimer as a coagulation factor and interleukin 6 as a proinflammatory cytokine that can induce coagulation, especially due to long-term cytokine storms. Here, researchers try to make a suggested scheme based on the review before (
Figure 1). One of the proposed mechanisms is the direct viral infection of endothelial cells that line blood vessels. This infection can cause endothelial dysfunction, leading to the release of procoagulant factors and impaired regulation of anticoagulant mechanisms. Additionally, the dysregulated immune response and cytokine storm can further promote a prothrombotic state
[33][70]. Other further studies are needed to assess the correlation between each criterion and alteration or dysregulation of the immune system.
Figure 1. Proposed schematic for correlation between hepatocyte growth factor (HGF) with D-dimer and Interleukin 6 (IL-6) as a prognostic marker of coagulation and inflammation on long-term effects of COVID-19 survivor
[16][22][25][33][34][35][36][37,46,47,56,70,71,72].
In the proposed schematic (
Figure 1), infection from SARS-CoV-2 can trigger an inflammatory process in the body
[34][37]. The inflammatory process during SARS-CoV-2 infection is induced by monocytes and macrophages
[16][56]. The induction of monocytes and macrophages triggers the emergence of proinflammatory cytokines, especially IL-6
[16][56]. Proinflammatory cytokines induce proHGF secretion by stromal cells, which are then activated by Tissue Factor to become HGF
[25][47]. HGF regulates the upregulation of pro-coagulation factors (PAI-1&COX-2), which trigger disseminated intravascular coagulation (DIC)
[22][46]. IL-6 cytokines induce Tissue Factor
[35][71] and cause endothelial injury
[36][72]. The occurrence of DIC causes hypercoagulation and increases the concentration of D-dimer
[34][37]. The occurrence of endothelial injury causes platelet adhesion, thereby activating the coagulation cascade
[37][73]. Hypercoagulation causes high D-dimer concentrations, thereby increasing IL-6 concentrations
[35][71].