Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Amol Tandon and Version 2 by Camila Xu.
MYC deregulation, a cardinal event in Burkitt lymphoma (BL) pathogenesis, necessitates the elucidation of the molecular mechanisms governing MYC activation to devise innovative and effective therapeutic strategies.
- MYC
- MYC translocation
- MYC deregulation
- Burkitt lymphoma
1. Introduction
Burkitt lymphoma (BL) is a highly aggressive and rapidly advancing subtype of non-Hodgkin’s lymphoma, hallmarked by elevated proliferation rates and substantial genetic instability. Burkitt lymphoma (BL) has several notable distinctions in cancer research. It was the first human tumor found to be linked with a virus [1] and one of the earliest tumors identified to have a chromosomal translocation activating an oncogene [2]. Additionally, it was the first lymphoma reported in association with HIV infection [3]. Moreover, it was the first pediatric tumor to show a response to chemotherapy alone [4]. In regions with endemic malaria, such as equatorial Africa, Brazil, and Papua New Guinea, BL is the most prevalent childhood cancer [5][6][5,6].
A defining feature of BL is the presence of chromosomal translocations involving the MYC gene, which encodes a pivotal transcription factor that orchestrates cell growth, differentiation, and apoptosis [7][8][7,8]. The MYC oncogene significantly contributes to many human cancers by regulating myriad genes. Its influence extends beyond cellular biology to impact host immunity and the tumor microenvironment [9][10][9,10]. It is activated through various methods—genetic, epigenetic, and post-translational—leading to altered expression, and thus influences different types of cancer [11]. Common genomic changes, such as gene amplification, chromosomal translocations, and mutations, can escalate MYC expression [12][13][12,13]. The gene further influences cancer cells by managing processes, such as growth, differentiation, and metabolism [14][15][16][14,15,16]. Although MYC expression is typically restrained in healthy cells, its heightened levels in cancer cells can trigger apoptosis. Nevertheless, its activation bypasses these physiological checks, thereby promoting cancer via several interconnected mechanisms [9][17][18][9,17,18]. The regulation of MYC and the diverse range of its targets is depicted in Figure 1. MYC overexpression in Burkitt lymphoma is commonly facilitated by chromosomal translocations, such as the MYC-immunoglobulin heavy chain (IgH) [19][20][19,20]. This translocation culminates in unrestrained MYC activation, fostering cell proliferation and impeding apoptosis through intricate and diverse mechanisms that entail interactions with a plethora of proteins and regulatory factors [21].
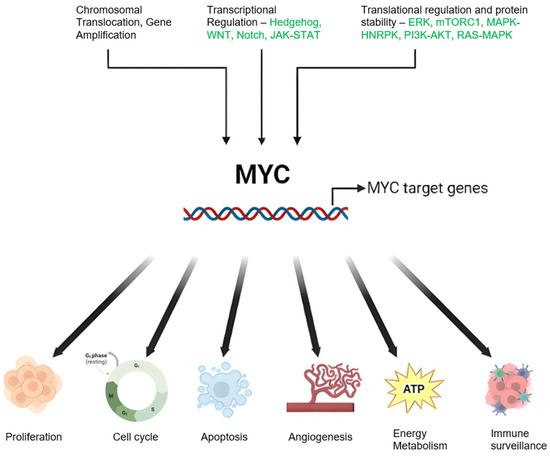
Figure 1. Schematic Representation of MYC Regulation and Function in Cancer: Genetic alterations including chromosomal translocations and genomic amplifications result in elevated MYC mRNA expression. Modifications in upstream regulatory pathways can also influence MYC oncogene transcription. MYC protein stability is enhanced through post-translational modifications, notably preferential phosphorylation at serine 62 (S62) over threonine 58 (T58), inhibiting degradation and stimulating the MYC pathway. MYC orchestrates a variety of cancer cell-intrinsic and host-dependent pathways to foster cancer cell growth and survival, with key processes including proliferation, metabolism, protein, and ribosomal biosynthesis. Conversely, MYC suppresses cellular protective mechanisms, such as immune surveillance, and thus aids in cancer progression. Notably, MYC can paradoxically trigger cellular processes, such as apoptosis that potentially threaten cancer cell survival. The ultimate cell fate is determined by the intricate equilibrium between these events and the specific cellular context. Created with BioRender.com, accessed on 20 May 2023.
In addition to chromosomal translocations, MYC point mutations have been identified in BL. Present in approximately 30% of BL cases [22], these mutations lead to increased MYC protein stability and activity, correlating with poor prognosis. Given the central role MYC occupies in BL development and progression, there is a burgeoning interest in devising MYC-targeted therapies. However, due to MYC’s transcription factor nature and the absence of a well-defined active region, the development of small compounds targeting MYC presents a formidable challenge [8].
2. MYC Deregulation in Burkitt Lymphoma: Mechanisms and Implications
- (1)
-
Chromosomal Translocations: In Burkitt lymphoma (BL), the most common mechanism of MYC deregulation involves chromosomal translocations that place the MYC gene under the control of immunoglobulin (Ig) enhancer elements, leading to constitutive activation of MYC expression in B-cells [23][24][25][26][34,35,36,37]. The translocation t(8;14)(q24;q32), found in approximately 80% of BL cases, is the most frequent MYC translocation in this disease [27][28][38,39]. Additional MYC translocations in BL include t(2;8)(p12;q24) and t(8;22)(q24;q11) [28][29][30][39,40,41]. Constitutive MYC overexpression drives uncontrolled cell proliferation, a defining feature of BL.
- (2)
-
Point Mutations: In addition to chromosomal translocations, point mutations in MYC have been identified in BL. The T58A mutation increases MYC protein stability by inhibiting its degradation via the ubiquitin–proteasome pathway and enhances MYC transcriptional activity by increasing its association with transcriptional co-activators, such as TRRAP and p300/CBP [31][32][42,43].
- (3)
-
Genomic Instability: MYC translocation also contributes to the genomic instability of BL cells. MYC-induced DNA replication stress can cause DNA double-strand breaks, leading to chromosomal rearrangements and mutations [33][34][44,45]. This instability can contribute to the clonal evolution of BL and the acquisition of additional mutations that drive tumor progression and resistance to therapy [35][46].
- (4)
-
Signaling Pathways: The molecular mechanisms of MYC-induced transformation in BL are complex and involve the deregulation of multiple signaling pathways. MYC regulates the expression of genes involved in several growth/proliferation regulating signaling pathways, including the Wnt/β-catenin, NF-κB, and PI3K/Akt/mTOR pathways [36][37][47,48]. Deregulation of these pathways can contribute to the aggressive behavior of BL and resistance to therapy.
- (5)
-
MYC and Cancer Metabolism: MYC has been implicated in cancer metabolism, particularly the Warburg effect, where it promotes glucose metabolism and aerobic glycolysis in cancer cells [38][39][49,50]. This metabolic adaptation enables cancer cells to withstand nutrient scarcity and hypoxic environments. MYC also plays a role in mitochondrial biogenesis and regulation in response to growth signals and cell cycle progression [40][41][42][51,52,53]. This provides an opportunity for therapeutic targeting of mitochondrial factors regulated by MYC in the context of the Warburg effect in cancer.
- (6)
-
MYC and Immune Regulation: Studies on human patient samples and transgenic mouse models have provided evidence that MYC is involved in the regulation of innate immune regulator CD-47 and the well-known adaptive immune checkpoint PD-L1 [43][44][45][54,55,56]. Saravia et. al [46][57] found that MYC also promotes the differentiation and activation of regulatory T-cells, which suppress immune responses and promote tumor growth. MYC influences the expression of genes involved in T-cell activation while suppressing the expression of genes involved in T-cell differentiation and function [46][57].
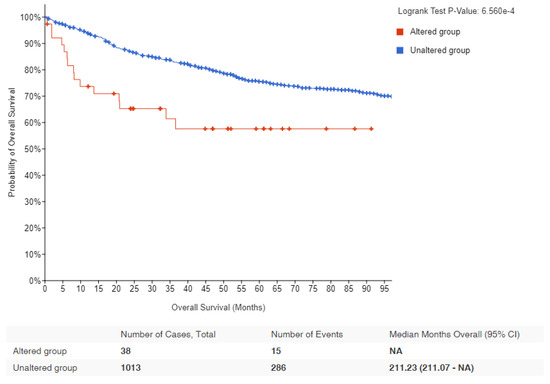 Figure 2. Kaplan–Meier survival estimates in lymphomas with MYC alteration: Data obtained from cBioPortal for lymphoma patients showing the probability of overall survival in MYC altered (red) and unaltered (blue) groups. Logrank p-value and number of samples analyzed for each group are indicated [47][48][58,59].Table 12.Summary of various mechanisms by whichMYCcould be deregulated.
Figure 2. Kaplan–Meier survival estimates in lymphomas with MYC alteration: Data obtained from cBioPortal for lymphoma patients showing the probability of overall survival in MYC altered (red) and unaltered (blue) groups. Logrank p-value and number of samples analyzed for each group are indicated [47][48][58,59].Table 12.Summary of various mechanisms by whichMYCcould be deregulated.Mechanism Description Gene Amplification This is when there is an increase in the number of copies of the MYC gene, leading to overproduction of MYC protein. Chromosomal
TranslocationThis is when the MYC gene is moved to a different chromosome, often leading to its inappropriate activation. In Burkitt lymphoma, for example, MYC is often translocated to the immunoglobulin heavy or light chain loci, which are highly active regions of the genome. Mutation Mutations in the MYC gene or in the genes that regulate MYC can lead to increased MYC activity. Deregulation of
Transcriptional ControlDisruptions in the normal mechanisms that control the transcription of the MYC gene can lead to overproduction of the MYC protein. Post-transcriptional and
Post-translational ModificationsChanges that occur to MYC mRNA or MYC protein after transcription and translation, respectively, can increase the stability, abundance, or activity of MYC. Deregulation of
MYC DegradationNormally, MYC protein is rapidly degraded to maintain proper control of its levels. Disruptions in these degradation pathways can lead to increased levels of MYC. 2.1. Mechanism of MYC Translocation and Its Significance in Accumulating Mutations on the Translocated Allele
2.1.1. Mechanism of IgH-MYC Translocation
Translocation occurs when chromosomes break and reattach to different chromosomes. In Burkitt lymphoma, one end of chromosome 8, where the MYC gene resides, translocates to chromosome 14, where the IgH gene is located. This leads to the fusion of the two genes. Normally, the MYC gene regulates cell division, but when it is fused with the IgH gene, it is then placed under the control of IgH’s regulatory elements. This results in overexpression of the MYC gene, leading to uncontrolled cell growth and proliferation, a key feature of lymphoma [11][49][11,60]. This translocation is often linked to errors in the somatic hypermutation process, a mechanism where B-cells modify their antibody genes to produce high-affinity antibodies. Normally, this process is tightly regulated, but errors can lead to off-target mutations, including the IgH-MYC translocation. In the context of chromosomal rearrangements leading to the translocation of proto-oncogenes into transcription-active regions, the study by Stasevich et al. [50][61] highlighted the role of non-coding enhancer RNAs (eRNAs) as regulatory mechanisms affecting the oncogenes upon translocation [50][61]. This study emphasizes the role of non-coding enhancer RNAs (eRNAs) in regulating the activity of oncogenes following translocation. The researchers sought to identify eRNAs that might influence MYC transcription during IgH-MYC translocation in Burkitt lymphoma, with an emphasis on eRNAs that are potentially oncogenic, located at the IgH locus, and predominantly expressed in B-cells. The results from this study demonstrated that eRNA AL928768.3 is a critical player in Burkitt lymphoma development. Importantly, the study’s findings also suggest that AL928768.3, along with other yet-to-be-discovered eRNAs, could potentially serve as tissue-specific targets for cancer therapeutics [50][61]. This opens up a promising new avenue for the development of targeted treatments for lymphomas, particularly Burkitt lymphoma.2.1.2. Risk Factors for Burkitt Lymphoma and IgH-MYC Translocation
Risk factors for Burkitt lymphoma, and thus the IgH-MYC translocation, include:2.1.3. Mutations Accumulate in the Translocated MYC Allele
The translocation of the c-MYC gene into the immunoglobulin heavy chain (IgH) locus in Burkitt lymphoma (BL) has been reported to result in a high number of mutations due to its exposure to the antibody hypermutation mechanism [54][55][56][57][58][65,66,67,68,69]. Bemark and Neuberger [54][65] demonstrated that the translocated c-MYC undergoes constitutive hypermutation in the Ramos BL cell line at a rate comparable to that of immunoglobulin variable (IgV) genes. This study found that the non-random distribution of mutations stems from the intrinsic bias of the mutational process, rather than a skewing effect of selection, and provided insights into the cis-acting sequences required for mutability. Specifically, IgH sequences downstream of the Sμ region were sufficient, and the IgH intronic enhancer was not necessary. Nearly two decades later, ref. [59][70] examined the effect of Phorbol 12-myristate 13-acetate (PMA) stimulation on the expression of the MYC gene in Ramos BL wild-type (WT) and ADP-dependent glucokinase (ADPGK) knock-out (KO) cells. The researchers discovered a larger accumulation of mutations in MYC transcripts in Ramos WT cells compared to ADPGK KO cells upon PMA stimulation. These mutations displayed a preference for AGC triplets, indicating the influence of immunoglobulin hypermutation and an organized mutational machinery. Interestingly, sequencing of stimulated Ramos BL cells revealed the recovery of the wild-type MYC sequence in nearly half of the cases, which contrasts the findings of [54][65]. Additionally, MYC expression increased during activation and decreased during differentiation as B-cells transitioned from an activated state to a plasma-cell-like differentiation phase, suggesting a functional role for MYC in providing energy stimulation to differentiating B-cells. Tandon et al. [59][70] also found that PMA stimulation increased MYC transcript levels in Ramos cells, but ADPGK KO cells showed a smaller elevation in MYC expression compared to WT cells. The study revealed that the accumulation of mutations is dependent on cell transcriptional activity rather than activation-induced cytidine deaminase (AID) expression levels. This finding points to a role for MYC in offering an evolutionary growth advantage to cancer cells in vitro, characterized by a higher rate of proliferation and increased transcriptional activity at the translocated MYC locus. In a separate study, ref. [60][71] investigated the mutational landscape of BL using high-throughput sequencing. They identified recurrent mutations in genes associated with the B-cell receptor signaling pathway, chromatin remodeling, and DNA damage response. This research not only provided valuable insights into the genetic mechanisms underlying MYC-driven lymphomagenesis in BL, but also highlighted potential therapeutic targets for future treatment strategies.2.2. Additional Genetic Alterations in Burkitt Lymphoma
Although MYC deregulation is a hallmark of Burkitt lymphoma (BL), it alone is insufficient to cause the disease. Additional genetic modifications are necessary for the transformation of B-cells into malignant lymphomas [61][72]. These additional mutations are generally thought to occur independently of the IgH-MYC translocation, namely, the IgH-MYC translocation does not directly cause these additional mutations to occur. However, it is worth noting that the overexpression of MYC might indirectly contribute to a higher mutation rate. MYC overexpression can lead to increased cell proliferation and DNA replication, which can increase the chance of errors occurring during DNA replication. Furthermore, MYC can induce a state of “replicative stress”, which can lead to DNA damage and genomic instability, further increasing the potential for additional mutations.2.2.1. TP53 Mutations and Its Prognostic Importance in BL
TP53 is a tumor suppressor gene that encodes a transcription factor that regulates cell cycle arrest, apoptosis, and DNA repair in response to cellular stress. TP53 is frequently altered in various cancers, including BL, where it is one of the most common and important co-alterations, but its role and impact on BL biology and clinical outcome are not fully understood [62][63][73,74]. Several studies have investigated the frequency, type, and prognostic value of TP53 abnormalities (mutations, deletions, and/or copy number neutral loss of heterozygosity) in BL, using different methods, such as sequencing, fluorescence in situ hybridization (FISH), immunohistochemistry, and gene expression analysis [62][63][64][73,74,75]. The reported prevalence of TP53 abnormalities in BL ranges from 20% to 70%, depending on the cohort, subtype, and detection method [62][63][64][73,74,75]. TP53 abnormalities are more common in adult than in pediatric BL, and in immunodeficiency-associated than in endemic or sporadic BL [28][39] TP53 abnormalities are also more frequent in cases with high-risk features, such as high lactate dehydrogenase (LDH), bone marrow or central nervous system involvement, or double-hit or triple-hit status (concurrent rearrangements of MYC, BCL2, and/or BCL6) [62][73]. In a comprehensive study conducted by Burkhardt et al. [65][76], they analyzed a large cohort of pediatric and adult Burkitt lymphoma (BL) cases. They discovered that the mutational profile of these cancers varies by age group. A critical finding was the significant prognostic impact of recurrent mutations in the TP53 gene, which are associated with diminished expression of TP53wt protein in lymphoma samples. This lack of TP53wt expression and presence of TP53 mutations were linked to decreased survival rates, suggesting the central role of TP53 in the disease’s progression. The study further implies that new therapeutic strategies could be developed to target this dysfunctional TP53, potentially by inhibiting its upstream regulators, MDM2 and MDM4. This could decrease the ubiquitination and subsequent proteasomal degradation of TP53wt. Additionally, the use of drugs, such as eprenetapopt, which have been shown to restore TP53wt levels, could be a promising approach [65][76] The identification of TP53 as a key molecular marker and driver of BL has important implications for diagnosis, prognosis, and treatment. TP53 testing should thus be incorporated into routine clinical practice to stratify patients according to their risk and response to therapy. Moreover, novel therapeutic strategies that target TP53 or its regulators may offer new opportunities to improve the outcomes of BL patients, especially those with relapsed or refractory disease. In addition to TP53 abnormalities, other factors that can affect the TP53 pathway in BL include the expression of MDM2 and MDM4, two negative regulators of TP53 that can bind to and inhibit its activity. MDM2 overexpression has been reported in some cases of BL, especially those with immunodeficiency or double-hit status [64][66][75,77]. MDM4 overexpression has also been identified as the only TP53 pathway abnormality in a subset of BL cases without TP53 or MDM2 abnormalities, suggesting a potential alternative mechanism of TP53 inhibition [64][75].2.2.2. TCF3 and ID3 Mutations in BL
Schmitz et al. [60][71] found mutations in transcription factor 3 (TCF3) in BL. TCF3 is expressed at high levels during B-cell development and is inhibited by the helix-loop-helix protein ID3. Another gene shown to be mutated in BL is cell cycle regulating Cyclin D3 (CCND3), which is a direct target of TCF3, refs. [60][67][68][71,78,79] confirmed these mutations in an independent study, analyzing their frequency and clinical relevance. Their findings revealed that CCND3 and ID3 mutations are correlated with an advanced stage of the disease, representing secondary hits after MYC deregulation, and are essential for lymphomagenesis. Mutations in ID3 have been identified in a subset of BL cases and are thought to contribute to the development of the disease by inhibiting B-cell differentiation and promoting cell proliferation [67][78] In a groundbreaking study, ref. [22] sequenced the genome of a BL tumor and the germline DNA from the same individual, as well as the exomes of over 50 BL tumors, comparing them to diffuse large B-cell lymphoma (DLBCL) tumor samples. They found differentially mutated genes in BL samples, such as ID3, PIK3R1, RET, GNA13, ARID1A, and SMARCA4. Importantly, they identified ID3 mutations occurring specifically in Burkitt lymphoma samples (34% of tumors) and not in DLBCLs. The study was the first to show that ID3 mutations are responsible for promoting cell cycle progression and proliferation in Burkitt lymphoma and identifying ID3 as a new tumor suppressor gene.2.2.3. MYC Translocation and 11q Alterations
Notably, MYC translocation serves as both a diagnostic hallmark and a critical prognostic factor for BL [69][80]. Patients afflicted with MYC-positive lymphomas face poorer prognoses and necessitate more aggressive treatment regimens compared to their MYC-negative counterparts [26][70][37,81] MYC-negative lymphomas, such as those harboring 11q alterations, are associated with a subtype of high-grade B-cell lymphoma, previously referred to as Burkitt-like lymphoma [71][82]. The lymphomas with 11q aberrations do not have the characteristic MYC translocations seen in Burkitt lymphoma. Instead, they exhibit chromosomal abnormalities involving the loss of a region on the long arm of chromosome 11 (11q). This region contains several tumor suppressor genes, such as ATM and BIRC3, which play roles in DNA repair and apoptosis. Loss of these tumor suppressor genes can contribute to the development of lymphoma. Although both MYC-positive and -negative subtypes share some genomic features, the presence or absence of MYC translocations and 11q alterations help in distinguishing these two distinct molecular subtypes of high-grade B-cell lymphomas. The grim prognosis of MYC-positive lymphomas (such as BL) is predominantly attributed to the intensified cellular proliferation and genetic instability driven by MYC’s activation of target genes governing cell cycle regulation, DNA replication, and DNA repair [72][83]. This genomic destabilization ultimately fosters the accumulation of additional mutations, accelerating disease progression and heightening resistance to treatment.2.3. Evolutionary Growth Advantage as an Implication of MYC Translocation in Cancer
Multiple studies have investigated the role of MYC translocation in providing an evolutionary growth advantage to Burkitt lymphoma (BL) cells. Mlynarczyk et al. [73][84] proposed that BL cells exploit the germinal center reaction to gain a competitive advantage. This complex process generates high-affinity B-cell receptors through somatic hypermutation and selection. MYC translocation drives the expansion of BL cells within the germinal center, promoting their survival and proliferation, facilitated by the interaction with the microenvironment, including T-cells and stromal cells [73][84] Epigenetic regulation of MYC in BL expression and progression has also been the focus of many studies [74][75][76][77][85,86,87,88]. Fernández-Serrano et al. [78][89] reviewed the role of epigenetic modifications, such as histone modifications H3K4me3 and H3K27ac, in regulating MYC expression in BL cells. Li et al. [79][90] demonstrated that MYC mutations in lymphomas are associated with changes in chromatin structure, increased levels of H3K27ac histone marks, and altered gene expression patterns, promoting lymphoma cell proliferation and survival. Cowling et al. [80][91] reported enhanced histone acetylation of MYC-target genes in lymphomas. Ref. [81][92] investigated the role of epigenetic mechanisms in MYC-driven lymphomagenesis, revealing alterations in DNA methylation patterns and their impact on specific genes involved in immunological regulation, cell adhesion, and cancer-related pathways; overall indicating a MYC-dominated growth advantage in these cells. Recent studies have also explored the critical role of microRNAs (miRNAs) and mutations in MYC and its translocated partner genes. Ref. [82][93] identified several microRNAs that regulate MYC expression, such as miR-150, miR-155, and miR-143, whose deregulation in BL leads to MYC overexpression. Several miRNAs, including miR-34a, miR-145, and the let-7 family, have also been shown to directly target MYC mRNA and negatively regulate its expression [83][94]. On the other hand, it has also been suggested that c-MYC modulates miRNA expression, revealing a complex interplay between c-MYC and miRNAs [84][85][95,96]. One study found 211 differentially expressed genes and 49 differentially expressed miRNAs in BL [86][97]. A significant finding was the downregulation of ATM and NLK genes, important regulators in response to DNA damage in BL tumor cells. These tumor suppressors were targeted by multiple upregulated miRNAs which could account for their aberrant expression in BL. The combined loss of p53 induction and function due to miRNA-mediated regulation of ATM and NLK, along with the upregulation of TFAP4, may be central for human miRNAs in BL oncogenesis. This allows for the survival of BL tumor cells with the IgH-MYC chromosomal translocation and promotes MYC-induced cell cycle progression, initiating BL lymphomagenesis [86][97]. Klanova and Klener [87][98] found that mutations in MYC-translocated partner gene, BCL2, promote the growth and survival of lymphoma cells by increasing the expression of MYC and other oncogenes. MYC mutations are also found to impact the tumor microenvironment, a determining factor for chemotherapeutic potency [88][89][90][99,100,101] These studies showed that MYC mutations in lymphomas lead to increased recruitment of immune-suppressive cells, promoting tumor growth, angiogenesis, and metastasis.