Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Nives Pondeljak.
Atopic dermatitis (AD) is predominantly characterized by eczema, dry skin, and itching. These symptoms are age-dependent and often associated with other atopic diseases (allergic asthma, allergic rhinitis, and food allergies).
- atopic dermatitis
- atopic eczema
- multidisciplinary
1. Introduction
Atopic dermatitis (AD) is predominantly characterized by eczema, dry skin, and itching (Figure 1) [1,2,3,4,5][1][2][3][4][5]. These symptoms are age-dependent and often associated with other atopic diseases (allergic asthma, allergic rhinitis, and food allergies). Numerous hereditary and environmental factors, and their mutual interactions, participate in the development and clinical manifestations of AD, which can vary significantly in appearance, intensity, and course. The latest findings regarding AD pathogenesis point to a disturbance in the function of the epidermal barrier, a disruption of the immune response, colonization of the skin by microorganisms, an increased tendency toward infection, and certain psychological factors among other causes/triggers.
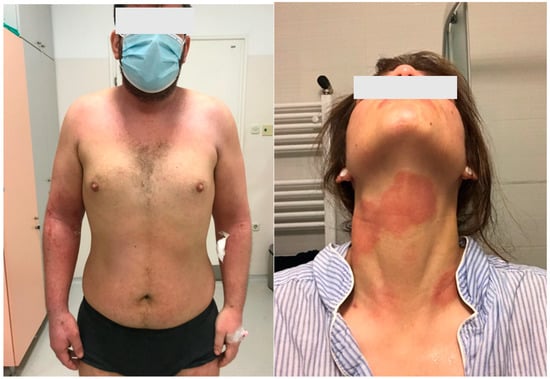
Figure 1.
Clinical pictures of the patients with atopic dermatitis.
The prevalence of AD has doubled or tripled over the last 30 years in industrialized countries. It is estimated that 15% to 30% of children, and 2% to 10% of adults, are affected [4,5,6,7][4][5][6][7]. AD typically occurs in the first year of life (in about 60% of cases) but may appear at any age, and its complete remission has been observed in 70% of children before adolescence. Early onset of the disease, familial associations, and early allergen hypersensitivity are all risk factors for a prolonged disease course and persistence into adulthood [8,9][8][9].
2. Basic Factors Involved in the Etiopathogenesis of Atopic Dermatitis
The etiopathogenesis of AD is complex and poorly understood, but it is thought to be caused by a complex interaction of genetic and environmental factors [1,4,10,11,12,13,14,15,16][1][4][10][11][12][13][14][15][16]. Thus, the pathogenesis of AD includes epidermal barrier dysfunction, immune dysregulation, and alteration of the skin microbiome, all of which can be altered by genetic and environmental factors. In particular, the key pathomechanism of AD development is the disruption of the epidermal barrier’s function and a changed immune response. Epidermal barrier dysfunction permits easier entry of microbes, allergens, and irritants, which trigger activation of immune response and release of pro-inflammatory cytokines. So, impairment of epidermal barrier function leads to immune dysregulation of innate and adaptive responses to environmental stimuli. It is known that the adaptive response is represented by Th2 cytokines, namely IL-4, IL-5, IL-13, and IL-31; eosinophilic activation; and production of allergen-specific IgE. In addition, disruption of the epidermal barrier’s function is related to the filaggrin protein, which is, along with keratin, the most important structural protein of the stratum corneum. A mutation of the filaggrin gene on chromosome 1q21 is common in AD patients, and it is significantly associated with disruptions to the structure and function of the epidermal barrier. This leads to an increase in transepidermal water loss (TEWL) and decreased synthesis of natural moisturizing factors (NMFs), which are produced in the process of filaggrin deamination [10,11][10][11]. In addition, the pH of AD patients’ skin is increased, and this increase in skin pH stimulates the activity of serine protease and kallikrein, which weakens the action of the enzymes responsible for ceramide synthesis. Decreased ceramide production is also caused by increased sphingomyelin deacylase activity—high expression of sphingomyelin deacylase indicates ceramide deficiency—and ultimately improves the possibility of environmental allergens penetrating deep into the damaged epidermal barrier [11,12,13][11][12][13].
In the epidermal skin barrier of AD patients, there is a correlation between immunological and structural disturbances. Genetic factors play a role in the development of immune barrier dysfunction, specifically chromosome 5q31-33 as it contains a group of family genes that code for the synthesis of Th2 cells that secrete the interleukins IL-3, IL-4, IL-5, and IL-13 and granulocyte-macrophage colony-stimulating factor (GM-CSF), which are important in the inflammatory response [14,15,16][14][15][16]. The deficiency of filaggrin increases the expression of keratinocyte-derived thymic stromal lymphopoietin (TSLP), which is crucial for the onset of skin inflammation in AD. So, TSLP activates dendritic cells that stimulate inflammatory Th2 cell differentiation and cytokine production. It seems that in AD patients, the increased Th2 cytokine expression causes an increase in serine proteases. In addition, tumor necrosis factor (TNFα) together with Th2 cytokines (IL-4, IL-13, IL-31) enhances the release of TSLP and limits the synthesis of long-chain free fatty acids, which allows damage to the skin’s lipid barrier to occur [12]. In addition, interferon-γ (IFN-γ) is involved in keratinocytes’ release of cytokines and chemokines, as well as IL-31, which exacerbates itch and is associated with mechanical trauma (e.g., the impulse to scratch the skin) [17,18][17][18]. In addition, exposure to environmental allergens and bacterial infections significantly increases the expression of TSLP, IL-25, and IL-33 and enhances the Th2-dependent response that triggers various interrelated events, leading to a chronic and recurrent AD course [19].
The inflammatory response of AD patients is mainly associated with the activation of T cells, dendritic cells, keratinocytes, macrophages, mast cells, and eosinophils. In acute AD lesions, the infiltrate predominantly consists of CD41+ cells, antigen-presenting cells (APCs) (Langerhans cells, other dendritic cells, and macrophages) with IgE bound to surface receptors. Thus, in the dermis, T cells predominate, with an increased number of CD4+ cells [20]. On the other hand, with chronic lesions, there are considerable dermal accumulations of collagen with a decreased T-cell count and a rich cellular infiltrate of eosinophils and macrophages. In inflammatory skin lesions, the T cells stimulate effector cytokines and induce keratinocyte activation and apoptosis. In the peripheral blood vessels of AD patients, we see natural regulatory T cells (Tregs) (CD4+, CD25+, FoxP3+) with regular immunosuppressive activity allowing Tregs to interfere with Th1 and Th2 responses. Mutations of the nuclear factor of Tregs cause immune dysregulation characterized by increased IgE levels, food allergies, and eczema. According to research data on the role of Tregs in AD, after stimulation with staphylococcal superantigen (enterotoxin type B), Tregs lose their immunosuppressive activity, which can exacerbate skin inflammation [13].
A damaged epidermal barrier increases the local expression of pro-inflammatory cytokines and chemokines, which bind to specific vascular endothelial receptors, and this facilitates the migration of pro-inflammatory cells that form AD skin infiltrate [21]. Thus, impaired stratum corneum is associated with filaggrin and other barrier protein deficiencies, elevated levels of endogenous serine proteases, and abnormal lipid composition which leads to increased stratum corneum permeability. All of these lead to impairment of epidermal barrier function and a vicious cycle of immune dysregulation. In inflammatory AD lesions (acute and chronic), a significant increase in IL-4, IL-5, and IL-13 can be observed. Even in apparently healthy skin, an increased expression of IL-4 and IL-13 (Th2 cytokines) is seen, which is not true for other cytokines such as IL-5 or IFN-gamma. Some of the above-mentioned pro-inflammatory cytokines, in combination with costimulatory molecules, initiate the differentiation of B cells into plasma cells that produce IgE antibodies. In addition, in AD lesions, receptors for IgE have been detected on the surface of myeloid dendritic cells, i.e., Langerhans cells and inflammatory dendritic epidermal cells (IDECs). The IDEC cells are responsible for synthesizing and releasing Th1 cytokines, while Langerhans cells are crucial for triggering the allergic immune response and controlling the antigen presentation to T cells, which in turn migrate into the epidermis and release numerous cytokines that induce keratinocyte apoptosis [19]. Stimulation of receptors on IDECs promotes the release of many inflammatory factors, which contribute to the amplification of the immune response. In skin lesions of AD patients, the expression of IL-31 is markedly increased.
Chronic inflammation in AD is associated with the dominance of Th1 cytokines (IL-12, IL-18, IL-11, TGF-β, and IL-31). At the same time, IL-5 is responsible for the maturation of eosinophils and the prolongation of their survival. Aside from skin lesions/eczema, chronic inflammation includes, or can lead to, concomitant itch, caused by various factors such as IL-31 produced by Th2 cells, an increased histamine release, colonization of the skin by Staphylococcus aureus, and other secondary issues. Thus, IL-31 is a Th2 cytokine that is highly expressed in AD lesions. Exposure to a staphylococcal antigen leads to increased expression of IL-31, bridging a direct connection between staphylococcal exposure and pruritus. Staphylococcus aureus stimulates mast cell degranulation, histamine release, and Th2 inflammation. So, the Th2 response further damages the skin barrier, worsens itching, and facilitates dysbiosis in favor of Staphylococcus aureus. In addition, Langerhans cells and inflammatory epidermal dendritic cells with specific IgE attached to the high-affinity IgE receptor, along with dermal dendritic cells, capture allergens and antigens in the affected skin. This triggers an immune response that activates sensory nerves via cytokines such as IL-4, IL-13, and IL-31, resulting in itchiness. Generally, the main cytokines involved in the development of AD lesions are Th2 cytokines, primarily IL-4 and IL-13, and current therapy options are often based on their suppression. It is known that type 2 cytokines, IL-4 and IL-13, have a crucial role in Th2 cell differentiation, IgE production, and eosinophil recruitment. They contribute to itch and are considered relevant to chronic pruritus in patients with AD. The predominant effect of increased IL-4 and IL-13 expression is the recruitment of eosinophils and basophils to tissues, along with heightened activation of Th2 cells and increased production of IgE. Additionally, IL-4 plays a role in the activation of Th2 cells via JAK-STAT signaling, leading to the production of various cytokines and chemokines. Knowledge of these pathogenetic processes is crucial for understanding AD and the creation of more effective therapy options.
References
- Avena-Woods, C. Overview of atopic dermatitis. Am. J. Manag. Care 2017, 23, 115–123.
- Frazier, W.; Bhardwaj, N. Atopic dermatitis: Diagnosis and treatment. Am. Fam. Physician 2020, 101, 590–598.
- Torres, T.; Ferreira, E.O.; Gonçalo, M.; Mendes-Bastos, P.; Selores, M.; Filipe, P. Update on atopic dermatitis. Acta Med. Port. 2019, 32, 606–613.
- Bieber, T. Atopic dermatitis. Ann. Dermatol. 2010, 22, 125–137.
- Baron, S.E.; Cohen, S.N.; Archer, C.B. Guidance on the diagnosis and clinical management of atopic eczema. Clin. Exp. Dermatol. 2012, 37, 7–12.
- Silverberg, J.I. Public health burden and epidemiology of atopic dermatitis. Dermatol. Clin. 2017, 35, 283–289.
- Margolis, J.S.; Abuabara, K.; Bilker, W.; Hoffstad, O.; Margolis, D.J. Persistence of mild to moderate atopic dermatitis. JAMA Dermatol. 2014, 150, 593–600.
- Peters, A.S.; Kellberger, J.; Vogelberg, C.; Dressel, H.; Windstetter, D.; Weinmayr, G.; Genuneit, J.; Nowak, D.; von Mutius, E.; Radon, K. Prediction of the incidence, recurrence, and persistence of atopic dermatitis in adolescence: A prospective cohort study. J. Allergy Clin. Immunol. 2010, 126, 590–595.e1-3.
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: Part I. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 657–682.
- Irvine, A.D.; McLean, W.H.; Leung, D.Y. Filaggrin mutations associated with skin and allergic diseases. N. Engl. J. Med. 2011, 365, 1315–1327.
- Cork, M.J.; Danby, S.G.; Vasilopoulos, Y.; Hadgraft, J.; Lane, M.E.; Moustafa, M.; Guy, R.H.; Macgowan, A.L.; Tazi-Ahnini, R.; Ward, S.J. Epidermal barrier dysfunction in atopic dermatitis. J. Investig. Dermatol. 2009, 129, 1892–1908.
- Peng, W.; Novak, N. Pathogenesis of atopic dermatitis. Clin. Exp. Allergy 2015, 45, 566–574.
- Boguniewicz, M.; Leung, D.Y. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246.
- McAleer, M.A.; Irvine, A.D. The multifunctional role of filaggrin in allergic skin disease. J. Allergy Clin. Immunol. 2013, 131, 280–291.
- Thomsen, S.F.; Ulrik, C.S.; Kyvik, K.O.; Hjelmborg, J.v.; Skadhauge, L.R.; Steffensen, I.; Backer, V. Importance of genetic factors in the etiology of atopic dermatitis: A twin study. Allergy Asthma Proc. 2007, 28, 535–539.
- Ellinghaus, D.; Baurecht, H.; Esparza-Gordillo, J.; Rodríguez, E.; Matanovic, A.; Marenholz, I.; Hübner, N.; Schaarschmidt, H.; Novak, N.; Michel, S.; et al. High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat. Genet. 2013, 45, 808–812.
- Furue, M.; Furue, M. Interleukin-31 and Pruritic Skin. J. Clin. Med. 2021, 10, 1906.
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2021, 48, 130–139.
- Leung, D.Y.M. New insights into atopic dermatitis: Role of skin barrier and immune dysregulation. Allergol. Int. 2013, 62, 151–161.
- Agrawal, R.; Wisniewski, J.A.; Woodfolk, J.A. The role of regulatory T cells in atopic dermatitis. Curr. Probl. Dermatol. 2011, 41, 112–124.
- Sroka-Tomaszewska, J.; Trzeciak, M. Molecular mechanisms of atopic dermatitis pathogenesis. Int. J. Mol. Sci. 2021, 22, 4130.
More